Author: Denis Avetisyan
Researchers have developed a visual analytics system to help make sense of the complex movements of atoms during molecular simulations.
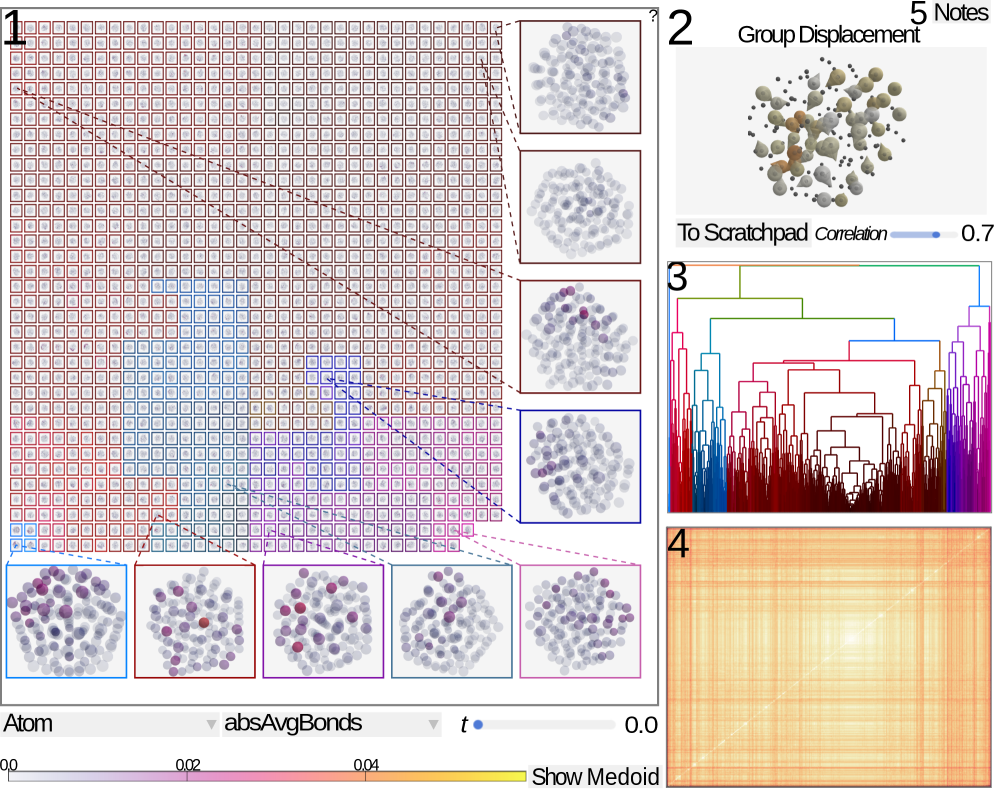
LAMDA facilitates the exploration and analysis of atomic displacements in molecular dynamics simulations using interactive visualizations and dimensionality reduction techniques.
Analyzing the atomic-scale evolution of materials via molecular dynamics simulations generates vast datasets, yet extracting meaningful patterns from these complex transitions remains a significant challenge. This paper introduces LAMDA: Aiding Visual Exploration of Atomic Displacements in Molecular Dynamics Simulations, a visual analytics system designed to facilitate systematic exploration of state-to-state atomic transitions through hierarchical categorization and interactive visualization. By enabling scientists to navigate and compare displacement behavior at multiple resolutions, LAMDA streamlines analysis and supports annotation-driven insights. Could this approach unlock a deeper understanding of material properties and accelerate the discovery of novel materials?
Decoding Atomic Motion: Navigating the Complexity of Transition Ensembles
Molecular dynamics simulations are increasingly utilized to model atomic rearrangements within materials, generating massive datasets known as ‘transition ensembles’. These ensembles capture the numerous pathways atoms take during a process-like a phase change or a chemical reaction-but their sheer size presents a significant computational hurdle. Analyzing these datasets isn’t simply a matter of increased processing power; the complexity arises from needing to discern meaningful patterns within a multitude of atomic configurations. Each transition represents a potential rearrangement, and the number of these transitions can quickly reach into the thousands, demanding innovative analytical approaches to filter noise, identify dominant pathways, and ultimately, connect these atomic-scale events to macroscopic material properties. The challenge lies not in generating the data, but in efficiently extracting knowledge from the vastness of atomic motion represented within these ensembles.
The sheer volume and intricacy of atomic rearrangements, as revealed by molecular dynamics simulations, present a significant hurdle for conventional visualization methods. Representing these ‘transition ensembles’ – sometimes encompassing up to 1400 distinct atomic pathways – quickly overwhelms traditional techniques, obscuring crucial details about material behavior. This difficulty isn’t merely aesthetic; the inability to effectively parse these complex datasets directly impedes the identification of dominant mechanisms, rare events, and subtle correlations that govern a material’s response to external stimuli. Consequently, advancements in visualization are essential not simply to show atomic motion, but to enable a deeper, more nuanced understanding of the fundamental processes driving material properties.
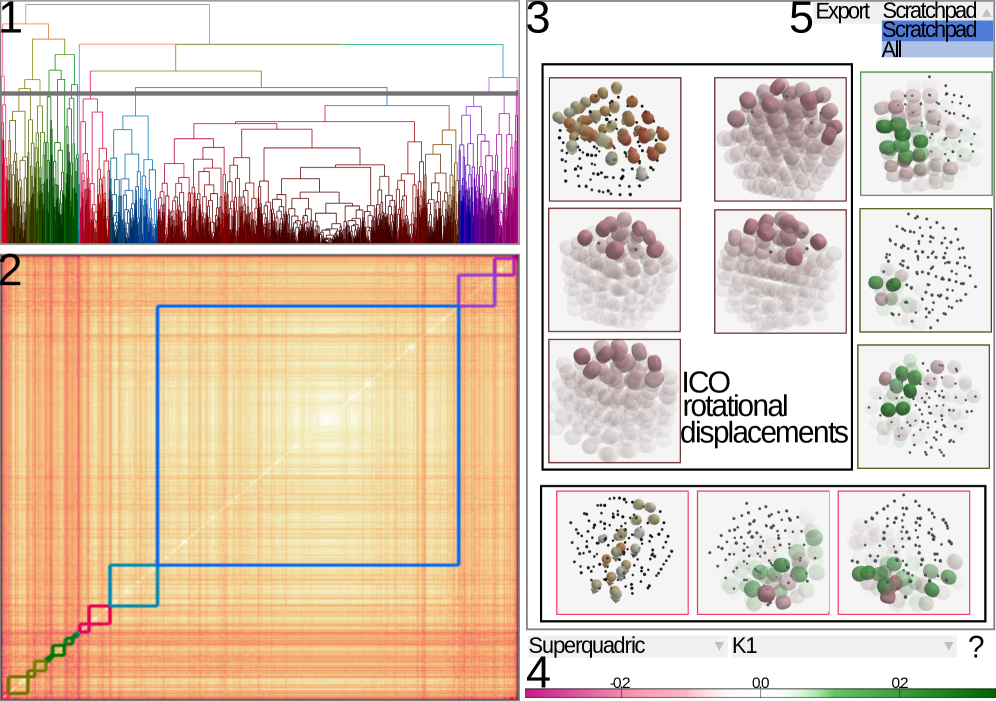
LAMDA: A System for Dissecting Atomic Rearrangements
LAMDA is a visual analytics system developed for the investigation of transition ensembles produced by molecular dynamics simulations. The system is designed to handle datasets containing a significant number of transitions, with demonstrated interactive performance on ensembles of up to 1400 transitions. This capability allows researchers to explore complex molecular rearrangements and identify patterns within large simulation outputs. LAMDA’s architecture is specifically tailored to manage the data volume and complexity inherent in analyzing multiple transition pathways simultaneously, enabling detailed investigation of atomic-level events.
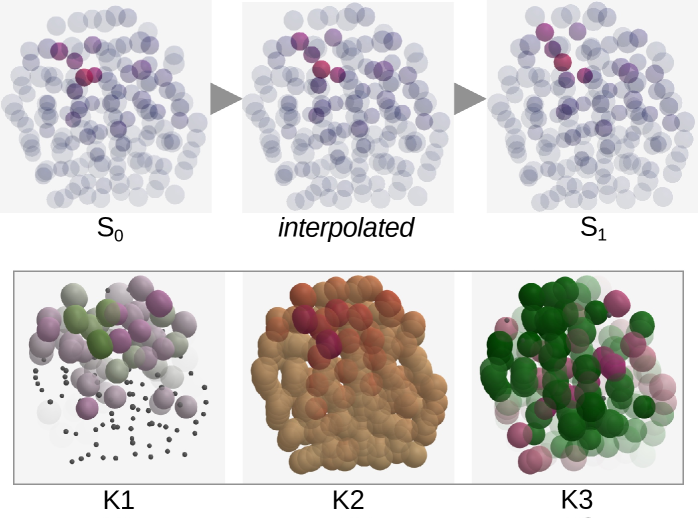
LAMDA employs a combination of visualization techniques to analyze atomic rearrangements within transition ensembles. Standard atom visualization provides a direct representation of atomic positions, while superquadric representations offer a simplified, abstracted view focusing on the overall shape and deformation of the molecular structure. Dendrograms are integrated to illustrate the relationships and branching patterns within the transition pathways, effectively clustering similar rearrangements based on structural similarity. This multi-faceted approach allows researchers to examine rearrangements at varying levels of detail, from individual atomic movements to global structural changes and pathway classifications.
LAMDA’s integrated visualization techniques enable the identification of key transition pathways and structural motifs through correlated visual exploration. Specifically, the system allows users to simultaneously view atomic movements, superquadric representations of conformational changes, and dendrograms illustrating the relationships between transition states. This multi-faceted approach facilitates the detection of recurring structural elements – motifs – and the tracing of dominant routes – pathways – through the ensemble of transitions. The combined visualization aids in discerning patterns that would be difficult to identify using single visualization methods, enabling researchers to characterize the mechanisms driving molecular rearrangements.
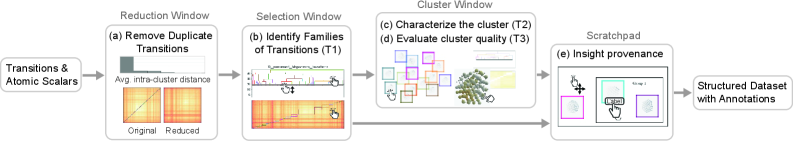
Refining the Signal: Reducing Noise in Transition Data
Transition reduction techniques are implemented to address the computational demands of large transition ensembles. This pre-processing step identifies and removes transitions exhibiting high similarity, thereby decreasing dataset size without significant loss of information content. Specifically, the application of these techniques resulted in an approximate 50% reduction in the number of transitions requiring further analysis. The methodology prioritizes the retention of transitions that represent distinct reaction pathways or structural configurations, ensuring the preservation of essential data for subsequent clustering and visualization processes.
Feature vectors are central to characterizing transitions by numerically representing the atomic environment at each transition state. These vectors are generated using techniques such as Common Neighbor Analysis (CNA), which identifies local structural motifs based on shared neighbors, and SNAP (Spectral Neighbor Analysis Potential) potentials, which utilize a Gaussian regression of atomic environments to determine potential energy. The resulting vectors quantify features like atomic density, bond angles, and coordination numbers, providing a basis for comparing and clustering similar transition states. Dimensionality reduction techniques are often applied to these vectors to improve computational efficiency and highlight the most significant features for clustering purposes, ultimately enabling the identification of distinct transition pathways and mechanisms.
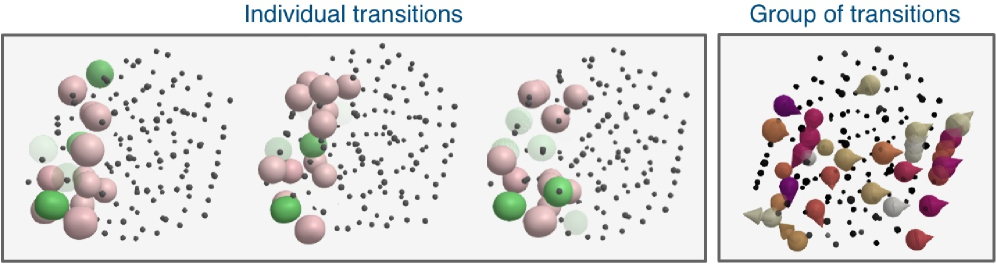
Following clustering of transition data, visualization employs group displacement methods to represent the collective movement of atoms during each transition. To enhance interpretability and facilitate spatial analysis, these clusters are then organized using a Hilbert space-filling curve. This curve effectively maps multi-dimensional data into a one-dimensional representation while preserving proximity relationships, allowing for improved visual arrangement and identification of trends. The complete pre-processing pipeline, encompassing data loading, calculation of invariants, and distance matrix computation, requires approximately 4 minutes and 6 seconds to execute.
From Local Motion to Global Understanding: Unveiling Material Behavior
Local Atomic Motion Detection Analysis, or LAMDA, offers a powerful means of dissecting how materials deform at the atomic level. Traditionally, observing deformation involved averaging behavior across large volumes, obscuring crucial details about where and how changes originate. LAMDA overcomes this limitation by pinpointing atomic-scale movements, allowing researchers to distinguish between alterations occurring at a material’s surface – such as oxidation or corrosion – and those happening within its internal structure due to stress or strain. This differentiation is critical, as surface transformations and internal rearrangements represent fundamentally different deformation mechanisms with distinct implications for a material’s overall properties and longevity. By isolating these processes, scientists gain a more nuanced understanding of how materials respond to external forces, paving the way for the design of stronger, more durable, and more resilient materials.
The reliability of computational materials science hinges on verifying that simulations accurately reflect real-world behavior. Researchers accomplish this by meticulously evaluating ‘cluster quality’ – a measure of how well-defined and stable atomic groupings are within a material undergoing deformation. By linking these observed cluster patterns to known crystalline structures, such as face-centered cubic (FCC) or icosahedral arrangements, scientists can directly validate the accuracy of their simulations. Specifically, the presence or absence of expected clusters within these structures confirms whether the simulation is correctly capturing fundamental deformation mechanisms. This detailed analysis not only bolsters confidence in predictive modeling, but also reveals crucial structural features that govern a material’s response to stress, offering pathways to tailor material properties for specific applications.
The ability to dissect material behavior at both local and global scales, as revealed through advanced analytical methods, directly empowers material design strategies. Understanding how microscopic rearrangements contribute to macroscopic properties allows researchers to move beyond empirical trial-and-error towards rational design principles. This nuanced comprehension facilitates the optimization of existing materials for enhanced performance – increasing strength, ductility, or resistance to specific environmental factors – and, crucially, enables the creation of entirely new materials with pre-defined functionalities. From lightweight alloys for aerospace applications to biocompatible implants and high-efficiency energy storage solutions, the potential for tailoring material characteristics to meet specific engineering demands is significantly broadened, promising innovations across diverse technological fields.
The system detailed in this research exemplifies a pragmatic approach to understanding complex systems. It doesn’t simply present data; it actively invites interrogation. This mirrors the sentiment expressed by John von Neumann: “If people do not believe that mathematics is simple, it is only because they do not realize how elegantly nature operates.” LAMDA, by facilitating the visual exploration of atomic displacements – a core concept within the Molecular Dynamics simulations – allows researchers to effectively reverse-engineer the ‘elegance’ of these interactions. The ability to cluster and analyze these transitions isn’t about finding pre-defined answers, but about constructing an understanding through controlled ‘breaking’ of the system – testing boundaries and observing emergent behavior. It’s a process of intellectual disassembly, revealing underlying principles through interaction and observation.
Pushing Beyond the Visible
The presented system, LAMDA, offers a navigable space within the chaos of molecular dynamics. But any tool that purports to aid understanding implicitly acknowledges the inherent limits of current approaches. The very act of reducing dimensionality, of clustering atomic displacements, introduces a filter – a deliberate simplification of reality. The crucial question isn’t whether LAMDA visualizes data effectively, but what is lost in translation. To truly understand these transitions, one must ask: what anomalies does the clustering hide? What rare events are smoothed over in the pursuit of pattern recognition?
Future work shouldn’t focus solely on refining the visualizations or scaling to larger datasets. Instead, the field should grapple with the epistemological implications of these analytical tools. Can one ever truly know a complex system through its representations? The next generation of visual analytics should prioritize methods for explicitly revealing the uncertainties inherent in the analysis, for highlighting what remains unseen, and for enabling researchers to actively break the established patterns to expose hidden mechanisms.
Ultimately, the value of a system like LAMDA lies not in providing answers, but in generating better questions. It is a starting point, a provocation. The true test will be whether it inspires researchers to not simply observe the dance of atoms, but to actively disassemble the choreography, to challenge the underlying rules, and to rebuild a more complete – and perhaps unsettling – picture of molecular behavior.
Original article: https://arxiv.org/pdf/2601.09887.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Review: Final Fantasy Tactics: The Ivalice Chronicles (PS5) – Still the Benchmark for Turn-Based Tactics
- Mark Zuckerberg & Wife Priscilla Chan Make Surprise Debut at Met Gala
- Elon Musk’s Mom Maye Musk Shares Her Parenting Philosophy
- 10 Greatest Manga Endings of All Time
- The WONDERfools ending explained: What happened to the Child of Eternity?
- The Witcher 3 Officially Reveals Stunning New Ciri Figure Coming 2026
- Nippon Sangoku Is The Best New Post-Apocalyptic Anime of Spring 2026
- The Best Action Movie of 2022 Is Finally Streaming Free (Just in Time for the Sequel’s Release)
- FRONT MISSION 3: Remake coming to PS5, Xbox Series, PS4, Xbox One, and PC on January 30, 2026
- 5 Things Jujutsu Kaisen Modulo Failed to Answer
2026-01-18 03:51