Author: Denis Avetisyan
A novel computational framework combining artificial intelligence and materials science dramatically accelerates the discovery of inorganic electrides with unique electronic properties.
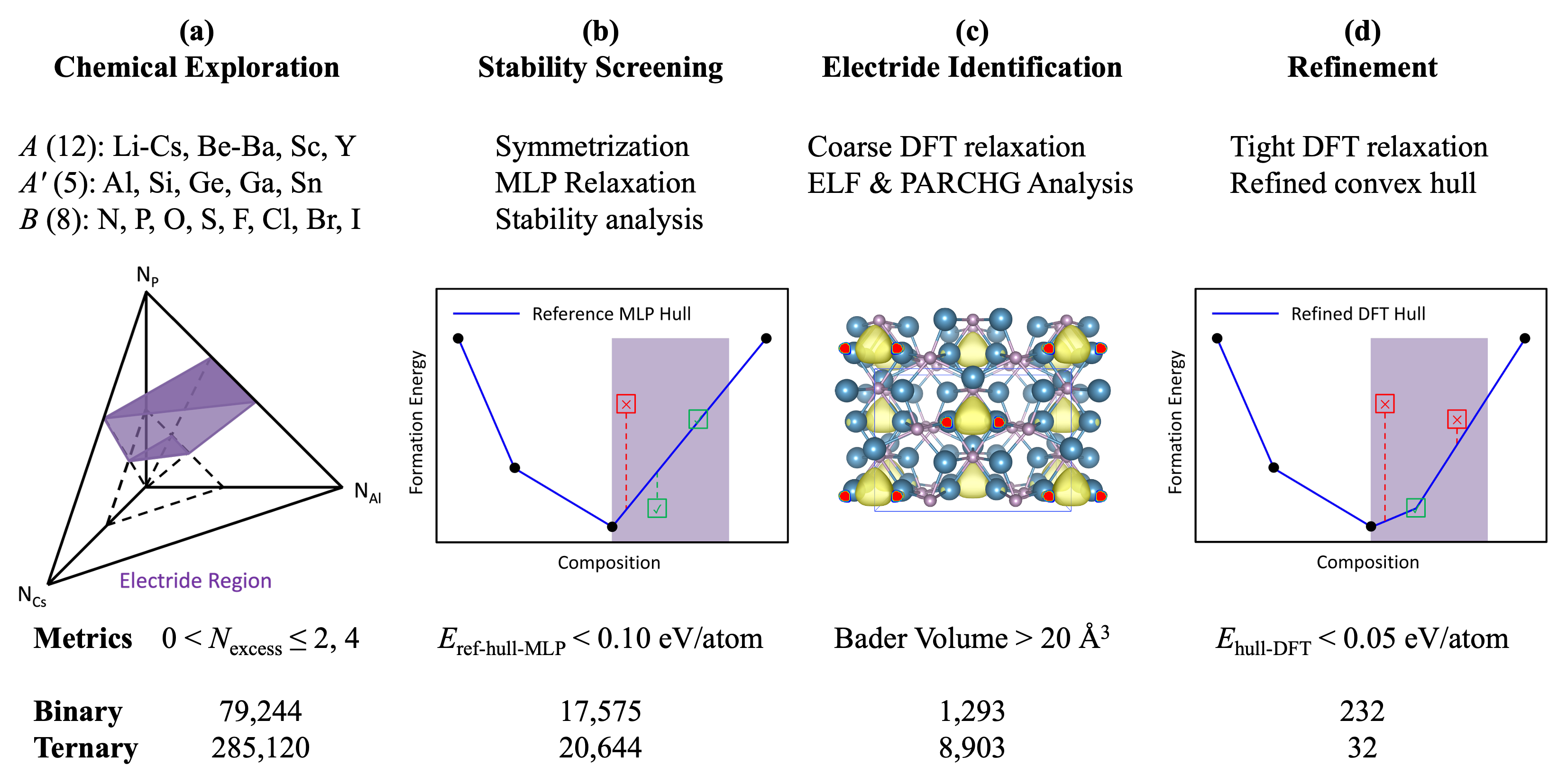
Researchers demonstrate a hierarchical screening process powered by generative models to identify 264 previously unknown inorganic electride candidates, significantly reducing computational cost and expanding the search for materials with interstitial electrons.
Despite their promise for applications ranging from catalysis to electron emission, discovering novel inorganic electrides-compounds hosting localized, interstitial electrons as anions-remains a formidable materials science challenge. In this work, ‘Accelerated Inorganic Electrides Discovery by Generative Models and Hierarchical Screening’ presents a computational framework that combines machine learning-driven materials generation with high-throughput thermodynamic and electronic structure screening to overcome this limitation. This approach efficiently explored a vast chemical space, identifying 264 previously unknown electron-rich compounds-including 13 predicted to be thermodynamically stable-and significantly reducing computational costs. Could this strategy unlock a new era of targeted materials discovery beyond electrides, rapidly accelerating the development of functional materials with tailored properties?
Unveiling the Electronic Landscape: Beyond Conventional Constraints
The functionality of most materials is constrained by the tightly bound nature of their electrons, limiting their potential in advanced technologies. Electrons in conventional solids typically occupy orbitals localized around atomic nuclei, restricting their movement and responsiveness to external stimuli. This inherent limitation hinders the development of materials with tailored electronic properties, such as enhanced conductivity, novel optical responses, or unconventional magnetic behavior. Consequently, researchers have increasingly sought materials where electrons are less confined, allowing for greater freedom and the emergence of previously unattainable functionalities. This pursuit has led to the exploration of materials like electrides, which challenge traditional constraints by hosting electrons in the space between ions, unlocking a pathway toward materials with exceptional and customizable properties.
Electrides are a fascinating departure from conventional ionic crystals, distinguished by the localization of electrons not within covalent or ionic bonds, but rather within the empty interstitial spaces between the constituent ions. This unique arrangement-where electrons effectively become anions-fundamentally alters the material’s electronic structure and gives rise to properties rarely observed in other compounds. These ‘excess electrons’ are not simply free electrons; they are weakly bound and highly mobile, contributing to exceptional electrical conductivity, high optical absorption in specific wavelengths, and even the potential for novel catalytic activity. Consequently, electrides present a compelling platform for developing materials with tailored functionalities, ranging from advanced energy storage to highly sensitive sensors and beyond, offering a pathway to technologies previously considered unattainable.
The defining characteristic of electrides – the presence of excess electrons not bound to individual atoms – dramatically reshapes the material’s electronic band structure. Unlike conventional materials where electrons primarily occupy states associated with atomic orbitals, electrides exhibit broadened and modified energy bands due to the delocalization of these interstitial electrons. This altered band structure isn’t merely a subtle shift; it fundamentally changes how the material interacts with light and electricity, potentially leading to extraordinary properties. Theoretical calculations and experimental observations suggest these excess electrons can contribute to enhanced conductivity, novel optical responses, and even the possibility of superconductivity at elevated temperatures. Furthermore, the unique electronic landscape of electrides allows for the creation of entirely new types of electronic devices and opens avenues for exploring previously unattainable phenomena in solid-state physics, promising a paradigm shift in materials science.
Accelerating Discovery: A Computational Lens on Electride Design
The conventional process of experimental materials discovery presents significant limitations in both time and resource allocation, especially when investigating complex compounds. Synthesizing and characterizing new materials requires iterative cycles of sample preparation, measurement, and analysis, often demanding months or years per material. This process is further complicated by the vast compositional space of potential materials, meaning researchers can only explore a tiny fraction of possible candidates. The cost associated with synthesizing, characterizing, and ultimately determining the stability of these materials is substantial, involving specialized equipment, skilled personnel, and significant consumption of resources. These factors collectively create a bottleneck in materials innovation, hindering the rapid development of materials with targeted properties.
An effective electride discovery process necessitates the integration of established physical principles with modern computational techniques. This workflow begins with defining relevant physical constraints, such as chemical composition and desired electronic properties, and then employs computational tools for structure generation and property prediction. Crucially, the workflow must include methods for assessing the thermodynamic stability of predicted structures, typically through techniques like the convex hull method, to filter out unrealistic or unstable configurations. The combined approach enables a systematic and efficient exploration of the vast chemical space, significantly accelerating the identification of potentially viable electride materials compared to purely experimental methods.
The computational workflow utilizes a generative AI model, MatterGen, to propose potential electride structures, coupled with machine-learned interatomic potentials implemented in MatterSim to efficiently calculate their energies and stability. This approach significantly accelerates the screening process; the generation of 79,244 binary structures required approximately 1000 GPU hours, representing a substantial reduction in computational expense compared to traditional methods. MatterSim’s machine-learned potentials enable rapid evaluation of a large chemical space, identifying promising candidates for further investigation, while MatterGen systematically explores structural variations to maximize discovery potential.
The thermodynamic stability of computationally predicted electride structures is rigorously assessed using the Convex Hull method. This technique involves calculating the total energy of a candidate structure and comparing it to the energies of its constituent binary end-members. The Convex Hull is constructed by plotting the energies of all possible binary compounds; a structure lying on the Convex Hull is considered thermodynamically stable, while those falling within the hull are metastable, and those outside are unstable. This analysis determines if a predicted electride is likely to exist under ambient conditions or if decomposition into more stable phases is probable, thereby filtering unrealistic structures from further consideration and validating the predictive power of the computational workflow.
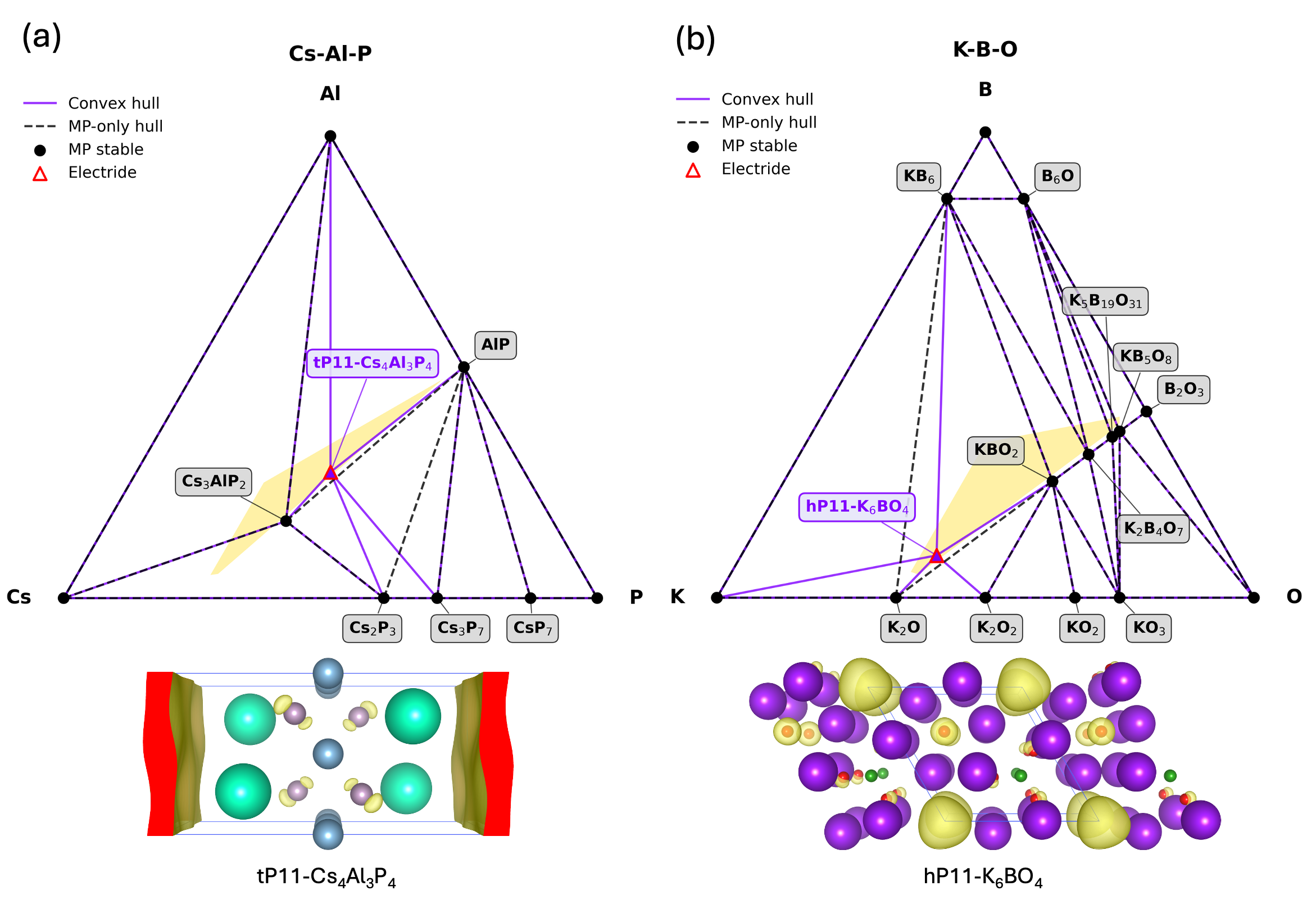
Expanding the Chemical Horizon: Beyond Binary Electrides
The computational workflow employed for electride discovery is not constrained to the investigation of binary compounds; it effectively integrates ternary compounds into the screening process. This expansion of the chemical search space is crucial, as it significantly increases the probability of identifying novel electride materials beyond the limitations of two-element systems. By incorporating ternary compositions, the workflow leverages a broader range of chemical bonding possibilities and electronic configurations, leading to the prediction of electride candidates that would be overlooked in binary-focused searches. This approach has proven successful in identifying a substantial number of new electride candidates, exceeding the number typically found through traditional binary compound investigation.
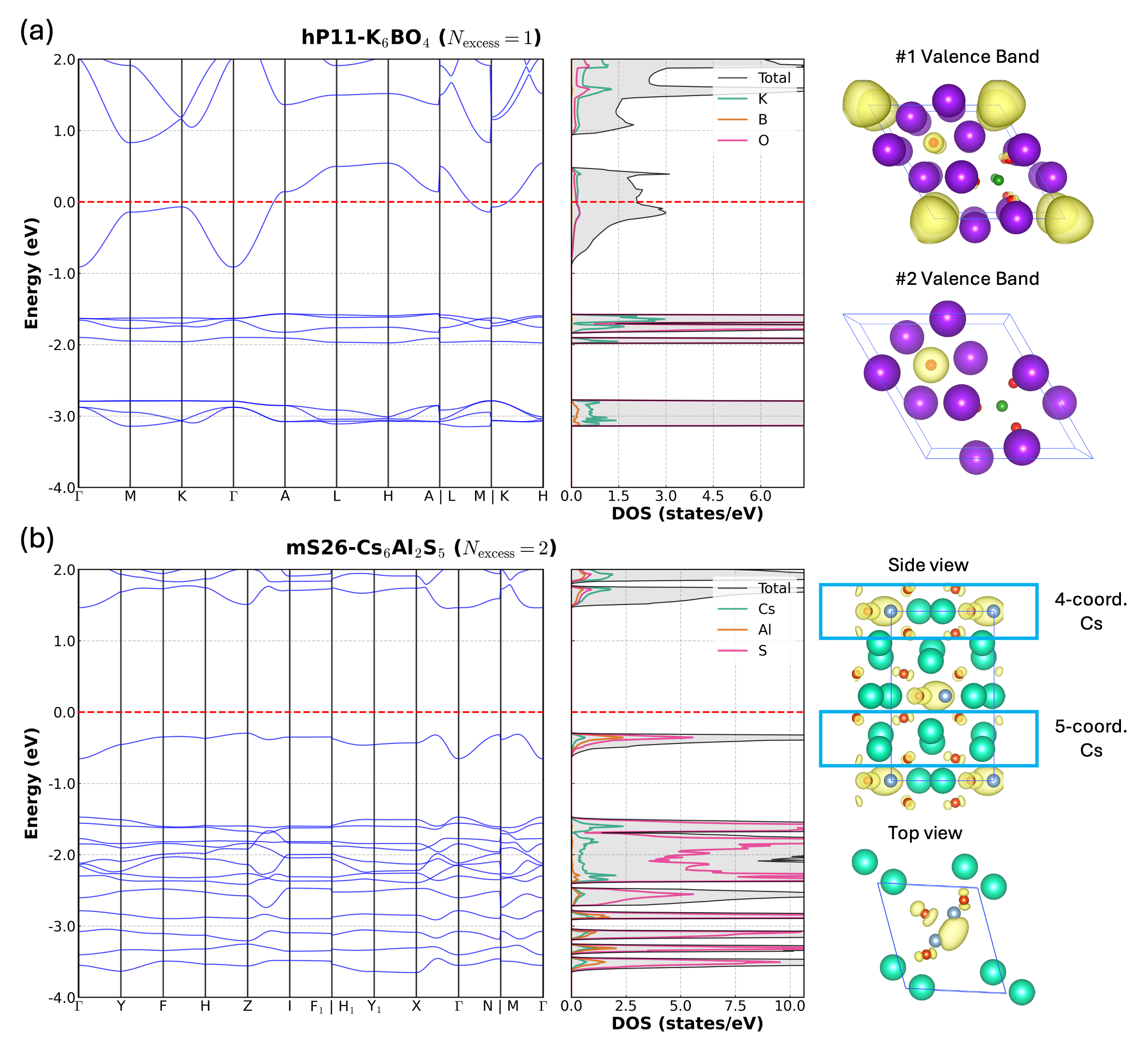
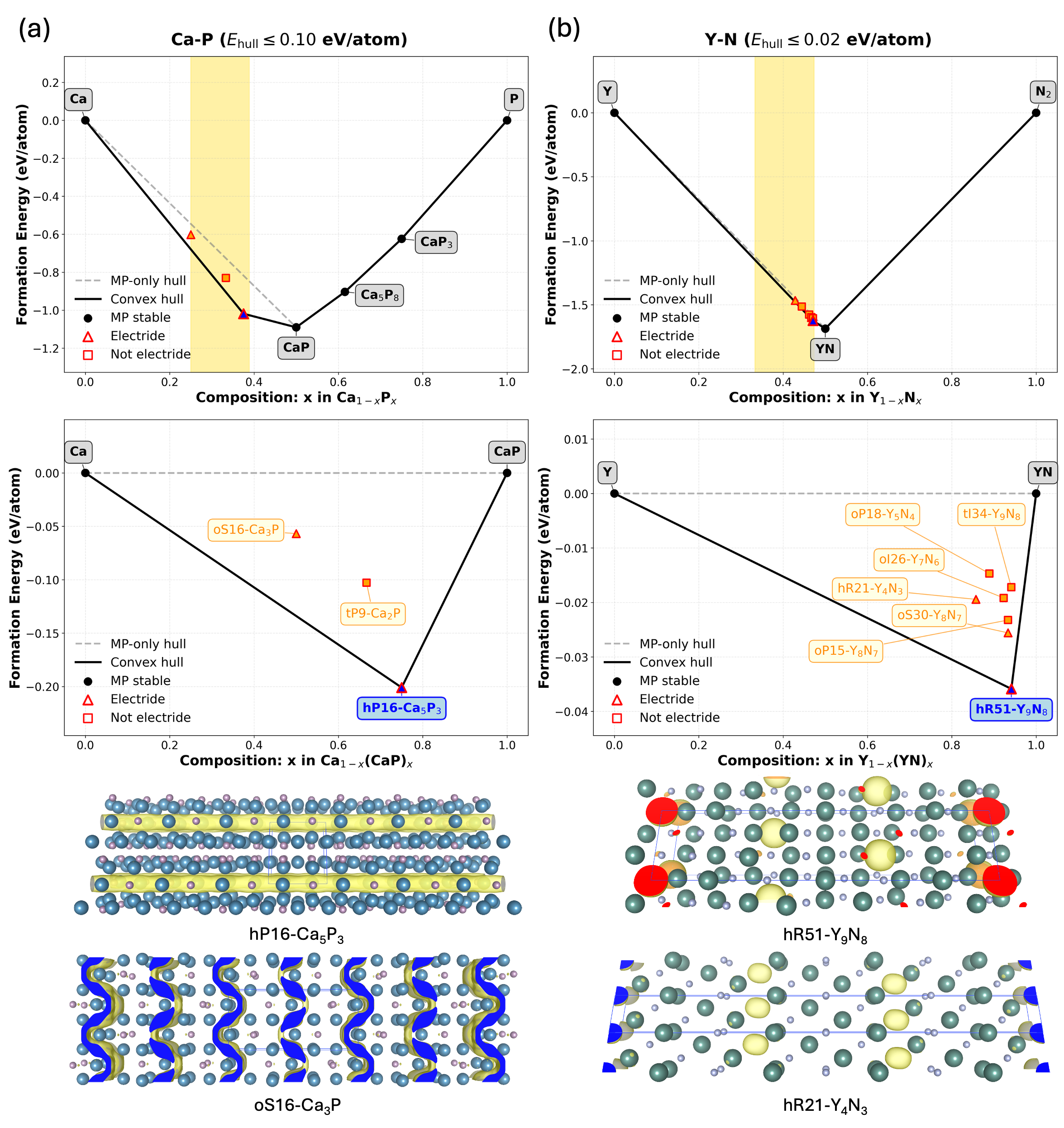
Computational predictions have yielded novel electride structures extending beyond previously known compositions. Specifically, one-dimensional electride materials with a Mn5Si3-type structure and two-dimensional electrides such as Ca2N have been identified. These materials exhibit characteristics indicative of potential applications, including high electron mobility and anisotropic magnetoresistance, validating the predictive capabilities of the employed computational workflow and demonstrating the expansion of the known electride material space to include these dimensionalities and compositions.
The computational workflow successfully predicted materials exhibiting properties desirable for electronic applications, notably high electron mobility and anisotropic magnetoresistance. This predictive capability resulted in the identification of 264 novel electride candidates, representing a substantial expansion of the known electride landscape. These predicted materials offer potential advancements in areas requiring efficient electron transport and magnetic responsiveness, and serve as a validation of the framework’s efficacy in discovering materials with targeted functionalities beyond previously known electride structures.
Initial electride candidates, including Cs+(18-crown-6)2e- and Ca6Al7O16 (C12A7:2e-), served as critical validation points for the predictive workflow by demonstrating the feasibility of identifying stable anionic electron states within complex compounds. Subsequent screening identified a total of 13 stable electride candidates, a determination supported by calculations revealing non-imaginary phonon frequencies across all candidate structures, indicating thermodynamic stability and structural integrity. These early successes established a benchmark for assessing the reliability of the predictive approach and its capacity to generate viable electride materials.
Towards Functionality and Innovation: The Future of Electrides
Electrides, materials characterized by the presence of freely mobile electrons residing in interstitial spaces, present a compelling pathway toward realizing topological insulating phases. This arises because the unique ‘Interstitial Electron Localization’ within the material’s structure fundamentally alters its electronic band topology. These localized electrons contribute to the formation of robust surface states – conducting pathways protected by time-reversal symmetry – which are hallmarks of topological insulators. By carefully controlling the composition and structure of electrides, researchers can tune the strength and distribution of these interstitial electrons, effectively ‘designing’ materials with specific topological properties. The resulting topological insulating phases hold immense promise for next-generation electronic devices, offering enhanced stability, reduced energy dissipation, and novel functionalities based on spin-polarized currents and quantum phenomena.
The realization of topological insulating phases in electrides, coupled with precise control over the material’s valence band structure, presents a pathway toward a new generation of electronic devices. By manipulating the energy and momentum of electrons within these bands, researchers can engineer materials exhibiting unique conductive properties – allowing current to flow along surfaces while remaining insulating in the bulk. This characteristic is crucial for developing low-power, high-efficiency transistors and spintronic devices, where electron spin, rather than charge, carries information. Furthermore, the ability to tailor these electronic bands offers the potential to create novel optoelectronic components, enhancing light-matter interactions and potentially leading to more sensitive sensors and advanced display technologies. This precise band engineering, facilitated by the unique properties of electrides, moves beyond conventional semiconductor limitations and unlocks possibilities for devices with unprecedented performance and functionality.
A streamlined computational workflow now offers materials scientists an unprecedented ability to design and discover novel electrides with pre-defined functionalities. This approach moves beyond serendipitous discovery, allowing researchers to virtually screen countless material compositions and crystal structures before committing to expensive and time-consuming synthesis. By integrating density functional theory calculations with automated high-throughput screening, the workflow efficiently predicts key properties – such as electron localization and band structure – guiding the search for electrides optimized for specific applications. This predictive power dramatically accelerates the materials discovery process, paving the way for rapid innovation in fields reliant on advanced electronic materials and enabling the creation of devices tailored to meet increasingly complex technological demands.
The continued investigation of electrides holds considerable promise for advancements across diverse technological landscapes. These materials, characterized by their unique excess electrons, are not merely a theoretical curiosity but a potential cornerstone for next-generation energy storage solutions, offering the possibility of significantly enhanced battery capacity and charging speeds. Beyond energy, the exotic electronic properties of electrides are increasingly recognized for their potential in quantum information processing, where their ability to host topologically protected states could lead to more stable and robust qubits. This convergence of materials science and quantum technology suggests that electrides may play a critical role in realizing fault-tolerant quantum computation, while also enabling the development of novel spintronic devices and highly sensitive sensors – fundamentally reshaping areas from data storage to medical diagnostics.
The accelerated discovery of inorganic electrides, as detailed in this work, echoes a fundamental principle of knowledge: understanding precedes creation. Each identified candidate material isn’t simply a novel structure, but a point within a complex chemical space revealed through systematic exploration. This methodical approach-leveraging generative models and hierarchical screening-uncovers structural dependencies otherwise hidden within vast datasets. As John Locke observed, “All knowledge is ultimately based on sensation.” In this context, ‘sensation’ translates to the data generated by computational methods, which, when rigorously analyzed, unveils the underlying principles governing material stability and the emergence of electrides. The efficiency gained through this framework allows researchers to move beyond trial-and-error, focusing instead on interpreting the models and validating hypotheses.
Future Directions
The identification of 264 novel inorganic electride candidates, while a substantial expansion of the known chemical space, merely sharpens the edges of the unknown. The current framework, predicated on density functional theory calculations, inherits the inherent approximations within those methods. The true test lies not in computational prediction, but in experimental validation – a process which often reveals the limitations of even the most sophisticated models. Discrepancies between predicted and observed behavior are not failures, but rather opportunities to refine the underlying physics and inform subsequent iterations of the generative model.
A compelling avenue for future work involves incorporating dynamic effects. The present study largely focuses on static structures, yet the behavior of interstitial electrons-and thus the functionality of these electrides-is likely sensitive to temperature, pressure, and external fields. Modeling these dynamic interactions presents a significant computational challenge, but promises a more nuanced understanding of electride properties. Further, extending the generative model to predict not just structure, but also synthetic accessibility, would greatly accelerate the translation of these computationally discovered materials into real-world applications.
Ultimately, the value of this work lies not in the number of candidates identified, but in the demonstration of a viable pathway for materials discovery driven by artificial intelligence. The model, as it stands, is a map, not a territory. Refining that map-correcting its distortions and expanding its scope-is the enduring task.
Original article: https://arxiv.org/pdf/2601.21077.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- What Song Is In The New Supergirl Trailer (& What It Means For The DC Movie)
- One of Hulu’s Best New Shows Lands on Disney+ Ahead of Season 2
- Eurogamer Gives ARC Raiders 2/5 Over AI Voices, Dropping Metacritic Score from 94 to 84
- Gold Rate Forecast
- Netflix’s Run Away is the latest show to fail the Gen Z social media test
- Meet the Real-Life Inspiration For Sex and The City’s Mr. Big
- Jujutsu Kaisen Is Returning With a Special Release After Sequel’s Finale
- The Quantum Observer: How Reality Takes Shape
- I Had High(ish) Hopes For Five Nights At Freddy’s 2, But My Two Most Glaring Issues Make Me Wary About A Third Movie
- Ariana Grande and More Celebs React to Golden Globes 2026 Nominations
2026-01-31 16:46