Author: Denis Avetisyan
Researchers have developed a new approach to training artificial intelligence models to accurately predict the transition states of chemical reactions, even for previously unseen molecules and complex systems.
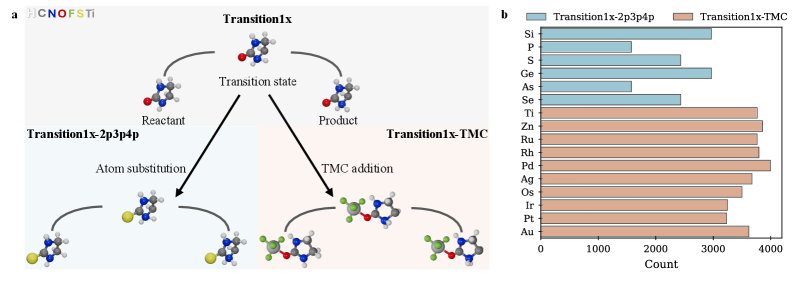
Self-supervised pretraining with equilibrium conformers enhances the generalization ability of generative models for transition state prediction, expanding the scope of computational chemistry.
Accurately predicting transition states is crucial for understanding reaction rates, yet current machine learning models struggle to generalize beyond their training data. In the work ‘Beyond the Training Domain: Robust Generative Transition State Models for Unseen Chemistry’, researchers address this limitation by introducing a self-supervised pretraining strategy using equilibrium conformers to enhance the generalization ability of generative models for transition state prediction. This approach substantially improves performance on reactions involving novel chemical elements and complex catalytic systems, reducing geometric errors and data requirements. Will this framework unlock efficient and scalable exploration of complex reaction landscapes beyond the limitations of traditional computational methods?
The Challenge of Transient States
The accurate modeling of chemical reactions relies heavily on identifying transition states – the fleeting, high-energy configurations representing the maximum energy point along a reaction pathway. However, locating these transition states presents a substantial computational challenge. Unlike stable reactants or products, transition states are saddle points on the potential energy surface, demanding sophisticated algorithms to pinpoint them without collapsing into minimization towards a local minimum. This computational expense scales rapidly with the size of the molecular system, effectively limiting the study of complex reactions, particularly those relevant to biological processes or materials science. Consequently, advancements in efficient transition state searching methods are crucial for accelerating chemical discovery and gaining a deeper understanding of reaction dynamics, as current limitations restrict simulations to smaller systems or require significant approximations.
Predicting the precise geometry of transition states – the fleeting structures between reactants and products – remains a substantial hurdle in computational chemistry, largely due to the demands of established methods. While techniques like Perturbation Theory with Reduced Functional Optimization (P-RFO) and Intrinsic Reaction Coordinate (IRC) calculations are known for their accuracy, they are notoriously computationally expensive. These methods require numerous energy and gradient evaluations, scaling poorly with system size and hindering their application to large, complex molecules or condensed-phase reactions. The computational cost becomes particularly prohibitive when exploring multiple possible transition states or when dealing with systems exhibiting significant conformational flexibility, effectively limiting the scope of chemical reaction modeling despite the reliability of the underlying principles.
Accelerating Discovery Through Generative Models
Generative models, including React-OT and AEFM, represent a shift from optimization-based transition state (TS) searching to direct prediction of TS geometries. These methods function by learning the potential energy surface (PES) of a chemical reaction from training data, enabling the generation of TS candidates without iterative minimization procedures. Unlike traditional approaches which navigate the PES to locate saddle points, generative models aim to directly sample geometries close to the TS, offering potential for significant computational speedups. The effectiveness of these models relies on their ability to accurately represent the complex PES and generalize to unseen chemical transformations, bypassing the need for expensive energy and gradient calculations at each step of the TS search.
Generative machine learning models accelerate transition state (TS) candidate generation by learning the potential energy surface (PES) of a chemical reaction. Traditional TS optimization algorithms iteratively refine a geometry until the minimum energy pathway is located, a computationally expensive process. These models, however, bypass iterative refinement by directly predicting likely TS geometries based on patterns discerned from the PES. This approach significantly reduces computational cost, as the model leverages learned relationships to propose viable candidates rather than relying on gradient-based searches. The efficiency stems from the model’s ability to approximate the PES and extrapolate to unseen chemical space, enabling rapid generation of TS structures for subsequent refinement or validation.
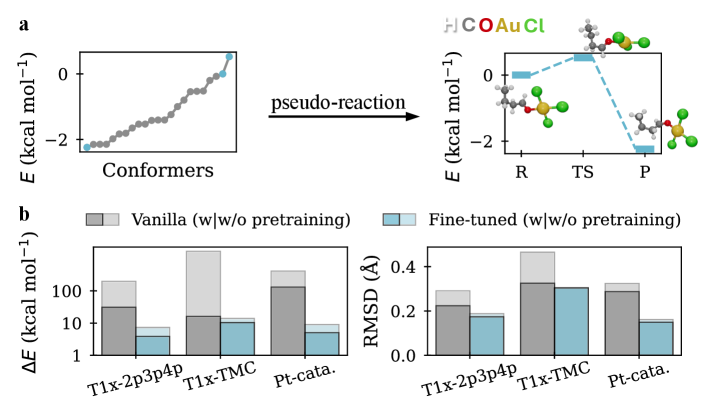
Pretraining generative transition state (TS) prediction models with pseudo-reactions derived from equilibrium conformer structures demonstrably enhances performance. This technique leverages a larger, synthetically generated dataset to improve the model’s understanding of chemical environments prior to optimization on actual TS data. Evaluation on the Transition1x-TMC benchmark indicates that models utilizing this pretraining approach achieve a median Root Mean Square Deviation (RMSD) of 0.19 Å for predicted TS geometries, representing a significant improvement over models trained without pretraining. The pseudo-reactions provide a broadened training scope, facilitating more accurate and efficient TS prediction.
Robustness and Generalization Assessed
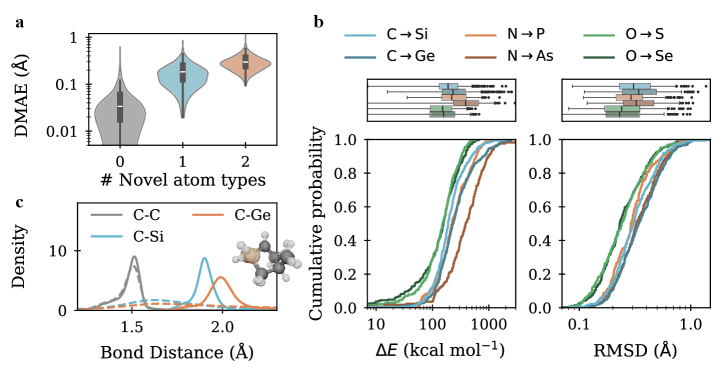
To comprehensively evaluate model generalization capabilities, the original Transition1x dataset was augmented with two new benchmarks: Transition1x-2p3p4p and Transition1x-TMC. Transition1x-2p3p4p introduces transition states containing second, third, and fourth-row elements, increasing the diversity of chemical species. Transition1x-TMC comprises transition states generated from the challenging TMC benchmark set, which features complex reaction mechanisms and steric interactions. These extensions were designed to move beyond the limitations of the initial dataset and assess performance on more varied and structurally demanding scenarios, providing a more robust evaluation of the model’s ability to extrapolate to unseen chemical transformations.
Model performance was evaluated using Root Mean Squared Deviation (RMSD), which quantifies the average distance between predicted and true transition state geometries; Discrete Manifold Alignment Error (DMAE), measuring the similarity of predicted and true structures after optimal superposition; and Wasserstein-1 Distance, assessing the distributional distance of predicted and true bond lengths. RMSD is reported in Ångströms (Å), providing a measure of geometric dissimilarity. DMAE focuses on the alignment of key structural features, independent of overall orientation. Wasserstein-1 Distance, also known as Earth Mover’s Distance, provides a metric for comparing the distributions of bond lengths, indicating how much “work” is required to transform one distribution into another.
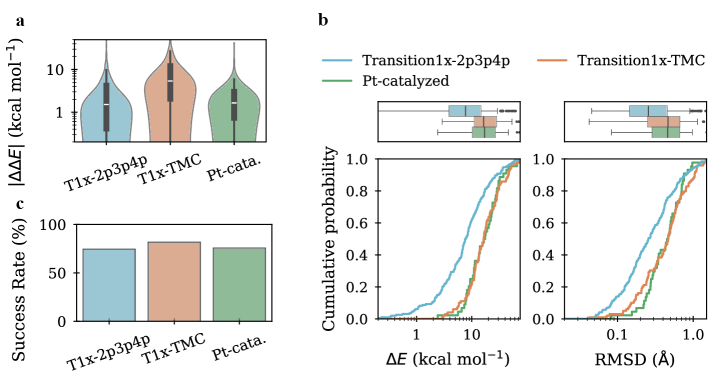
Following evaluation on the Transition1x-2p3p4p and Transition1x-TMC benchmarks, the models achieved a Root Mean Squared Deviation (RMSD) of 0.26 Å on Transition1x-2p3p4p and 0.42 Å on Transition1x-TMC. These values were obtained after Density Functional Theory (DFT) re-optimization of the generated transition state (TS) structures. This represents a significant performance increase compared to initial results, indicating improved accuracy in predicting TS geometries. The RMSD metric quantifies the average difference between predicted and true TS structures, with lower values indicating greater similarity and therefore, higher predictive power.
Self-supervised pretraining significantly improves data efficiency during model fine-tuning. Empirical results demonstrate a 75% reduction in the amount of labeled data required to achieve comparable performance when utilizing a self-supervised pretraining strategy. This indicates the model effectively learns transferable representations from unlabeled data, reducing its reliance on extensive labeled datasets during the supervised fine-tuning stage. The observed reduction in data requirements contributes to decreased computational costs and facilitates model application to scenarios with limited labeled data availability.
Towards Accelerated Chemical Design
The ability to swiftly predict transition states – the fleeting, high-energy configurations between reactants and products – is now being dramatically enhanced through innovative computational methods. This acceleration unlocks the potential for high-throughput screening, allowing researchers to systematically evaluate a vast landscape of reaction conditions and potential catalyst candidates. Instead of painstakingly analyzing each possibility, these techniques enable the rapid identification of optimal parameters for chemical reactions, significantly reducing the time and resources required for discovery. This approach isn’t limited to a single reaction; it facilitates the optimization of entire reaction networks, promising a paradigm shift in fields reliant on chemical synthesis, from the design of novel pharmaceuticals to the development of advanced materials with tailored properties.
The potential to drastically reduce the time required for chemical innovation promises a revolution across multiple scientific disciplines. Accelerated discovery cycles, facilitated by efficient computational methods, are poised to reshape drug development, allowing researchers to identify and optimize promising therapeutic candidates with unprecedented speed. Similarly, materials science will benefit from the ability to rapidly screen and design novel compounds with tailored properties, potentially leading to breakthroughs in areas like energy storage, sustainable polymers, and advanced manufacturing. This heightened efficiency isn’t merely incremental; it represents a paradigm shift, moving from largely empirical, trial-and-error approaches to data-driven, predictive modeling that substantially shortens the path from initial concept to tangible product. The convergence of computational power and innovative algorithms is thus unlocking a future where chemical discovery is limited primarily by imagination, not computational bottlenecks.
The convergence of machine learning with established quantum chemical calculations-such as Density Functional Theory (DFT) and the GFN2-xTB method-is forging a powerful new paradigm for chemical modeling. Rather than replacing computationally intensive yet highly accurate quantum methods, machine learning algorithms are being integrated to enhance them. These algorithms can be trained on data generated by DFT or GFN2-xTB to predict properties or reaction pathways with remarkable speed, effectively acting as a ‘force field’ that guides more precise, but slower, quantum calculations toward the most promising areas of chemical space. This synergistic workflow allows researchers to bypass the computational bottlenecks often associated with exhaustive quantum chemical searches, enabling the efficient exploration of complex chemical systems and accelerating the discovery of novel materials and pharmaceuticals. The resulting models offer a compelling balance between speed and accuracy, promising a future where complex chemical simulations are routinely performed with unprecedented efficiency.
The pursuit of predictive accuracy in complex chemical systems demands a parsimonious approach. This research, focusing on generative transition state models, embodies that principle. It streamlines the challenge of predicting reaction mechanisms by leveraging self-supervised learning from equilibrium conformers-a method of extracting signal from existing data rather than demanding exhaustive new calculations. As Max Planck observed, “A new scientific truth does not triumph by convincing its opponents and proclaiming its victories but by its opponents dying out.” The model’s ability to generalize to unseen chemistry suggests a similar dynamic; its structural honesty-rooted in efficient data utilization-renders alternative, more baroque approaches unsustainable. The focus on conformer sampling, a foundational element, emphasizes the value of simplicity in navigating complex conformational landscapes.
Where Do We Go From Here?
The pursuit of accurate transition state prediction remains, at its core, an exercise in controlled approximation. This work, by anchoring generative models in the readily available landscape of equilibrium conformers, represents a necessary subtraction – a move away from increasingly elaborate architectures seeking to solve chemistry, and toward systems that simply understand it a little better. The gains observed with novel elements and catalysts suggest the pretraining strategy addresses a fundamental limitation: the models were not learning chemistry, but memorizing training data. Yet, even a ‘chemically aware’ model is still a model.
The true test will not be reproducing known reactions, but predicting the unexpected. The current framework, while demonstrating improved generalization, still relies on density functional theory (DFT) for both pretraining data and evaluation. This circularity is not a failing, but a signpost. The next iteration must explore self-supervised learning loops that minimize reliance on external, potentially flawed, calculations. Can a model, given only the fundamental laws of physics and a corpus of molecular structures, begin to discover reaction mechanisms?
Ultimately, the goal isn’t a perfect model, but a minimal one. A system capable of identifying the essential degrees of freedom governing a reaction, and discarding the rest. This requires not more complexity, but a ruthless commitment to simplicity – a willingness to admit that most of what we calculate is, at best, noise. The path forward lies in embracing this uncertainty, and building models that are not defined by what they know, but by what they don’t need to know.
Original article: https://arxiv.org/pdf/2601.16469.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Dune 3 Gets the Huge Update Fans Have Been Waiting For
- Gold Rate Forecast
- Looks Like SEGA Is Reheating PS5, PS4 Fan Favourite Sonic Frontiers in Definitive Edition
- Pluribus Star Rhea Seehorn Weighs In On That First Kiss
- Kelly Osbourne Slams “Disgusting” Comments on Her Appearance
- Arknights: Endfield – Everything You Need to Know Before You Jump In
- Antiferromagnetic Oscillators: Unlocking Stable Spin Dynamics
- Tomodachi Life: Living the Dream ‘Welcome Version’ demo now available
- LeAnn Rimes Details “Craziness” of Affair With Eddie Cibrian
- ’90s Cartoon Reboot & TMNT Connection!
2026-01-26 22:20