Author: Denis Avetisyan
New research reveals that classical simulations struggle to reach conical intersections, fundamental to chemical reactions, due to an unexpected entropic barrier.
Molecular dynamics trajectories are prevented from reaching conical intersection seams, demonstrating a limitation of classical approaches to nonadiabatic dynamics.
Despite their central role in mediating nonadiabatic processes, conical intersections (CIs) are rarely, if ever, directly sampled in molecular dynamics simulations. This work, ‘The Entropic Barrier around the Conical Intersection Seam’, elucidates this behavior by demonstrating the emergence of an infinite free-energy barrier as the adiabatic gap closes at the CI seam. Using a linear vibronic coupling model and confirmed by simulations of methaniminium, we show that trajectories can approach regions of near-degeneracy but are fundamentally prevented from reaching the CI itself. Does this entropic barrier represent a universal constraint on nonadiabatic dynamics, and how might it inform the development of more efficient simulation methodologies?
Beyond the Limitations of Molecular Simulations
Molecular dynamics simulations, a cornerstone of computational chemistry, conventionally operate under the Born-Oppenheimer approximation. This principle drastically simplifies calculations by positing a clear distinction between the motion of electrons and nuclei within a molecule. Essentially, it assumes nuclei are stationary relative to the much faster-moving electrons, allowing for separate treatment of electronic and nuclear problems. This separation enables researchers to calculate potential energy surfaces upon which nuclear motion is then simulated. While remarkably successful for many systems, this approximation fundamentally relies on the premise that electronic and nuclear motions do not strongly influence each other, a condition that isn’t universally true and limits the accuracy of simulations when these interactions become significant.
The Born-Oppenheimer approximation, a cornerstone of molecular dynamics, falters when a system reaches a conical intersection – a point where multiple electronic states become indistinguishable. At these intersections, the usual separation between electronic and nuclear motion no longer holds, causing traditional simulations to fail and potentially yielding inaccurate results. This breakdown is particularly significant in photochemistry, where the absorption of light frequently drives molecules through these intersections, triggering reactions and structural changes. Consequently, a precise depiction of nonadiabatic processes – those involving transitions between electronic states – necessitates computational methods capable of handling the strong coupling between electrons and nuclei at and around conical intersections, ensuring a reliable understanding of complex photochemical events.
Accurately modeling nonadiabatic processes – those involving transitions between electronic states – demands computational approaches that move beyond treating electrons and nuclei as independent entities. Traditional methods often falter when these systems approach conical intersections, points where electronic states become closely coupled and the Born-Oppenheimer approximation fails. Consequently, researchers are developing techniques – such as surface hopping, multiple spawning, and exact propagation methods – that explicitly incorporate the interplay between electronic and nuclear degrees of freedom. These methods allow for the simulation of how a molecule’s potential energy surface changes as it traverses these intersections, capturing crucial dynamics for understanding phenomena like vision, photosynthesis, and the behavior of excited state molecules, ultimately providing a more complete picture of chemical reactivity and energy transfer.
A Mixed Quantum-Classical Approach to Nonadiabatic Dynamics
Mixed Quantum-Classical (MQC) simulations address the computational challenges inherent in modeling nonadiabatic dynamics by employing a partitioned approach. These methods leverage the differing timescales of electron and nuclear motion; electrons, exhibiting rapid behavior, are treated using quantum mechanical principles-typically described by the time-dependent Schrödinger equation-while nuclei, moving on slower timescales, are propagated classically using Newtonian mechanics. This partitioning significantly reduces computational cost compared to fully quantum dynamical treatments, enabling simulations of larger systems and longer timescales while retaining an accurate description of electronic transitions and their influence on nuclear motion. The validity of this approach rests on the Born-Oppenheimer approximation, where the electronic and nuclear wavefunctions are separable, and the assumption that nuclear motion does not significantly affect the electronic states during a single time step.
Trajectory Surface Hopping (TSH) is a Mixed Quantum-Classical (MQC) methodology for simulating nonadiabatic dynamics where trajectories representing nuclear motion evolve on a single potential energy surface (PES) until a hopping probability is calculated based on the gradients of the PESs and the energy gap between them. When a hop is accepted, the trajectory instantaneously switches to a different PES, representing a transition between electronic states; the hopping probability is formulated to satisfy the flux condition, ensuring conservation of probability during the simulation. This allows TSH to accurately model transitions between electronic states without requiring explicit solutions to the time-dependent Schrödinger equation for nuclear motion, making it computationally feasible for large systems where full quantum treatment is impractical.
Conical intersections (CIs) represent points in a potential energy surface where multiple electronic states become degenerate, facilitating nonadiabatic transitions crucial for understanding photochemical and photophysical processes. Simulations utilizing methods like Trajectory Surface Hopping, implemented in software packages such as SHARC 4.0, allow researchers to model molecular dynamics as trajectories evolve through these regions. These calculations provide insight into the rates and pathways of nonadiabatic transitions occurring at CIs, enabling the detailed investigation of phenomena like internal conversion and intersystem crossing. Specifically, SHARC 4.0 facilitates the simulation of systems with multiple coupled electronic states, tracking the probability of hopping between these states based on the Fermi’s Golden Rule and providing statistically significant data on reaction dynamics at conical intersections.
Unveiling the Entropic Barriers to Molecular Transitions
The S1/S0 conical intersection in the methaniminium cation (CH_2NH_2^+) presents a kinetic challenge beyond simple energetic considerations. While the potential energy surface allows for nonadiabatic transitions between the first singlet state (S1) and the ground state (S0) at the conical intersection, the system is hindered by an entropic barrier. This barrier arises from the zero-point vibrational energy and quantum tunneling effects, effectively restricting access to the minimum-energy region of the conical intersection seam. Consequently, the rate of nonadiabatic transitions is significantly reduced as classical trajectories are unable to surmount this barrier, and the system requires substantial vibrational energy to overcome the entropic penalty associated with approaching the conical intersection.
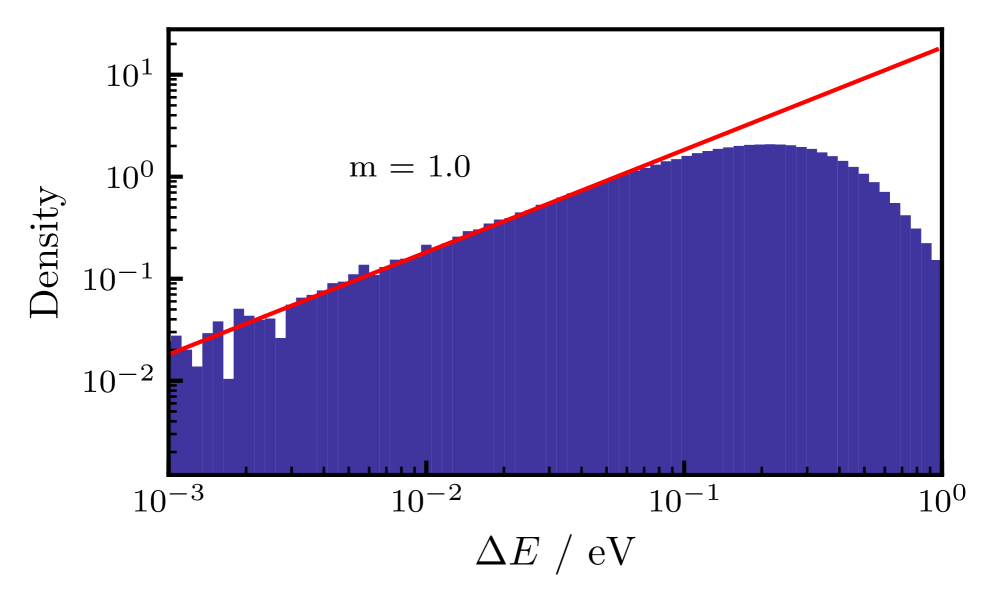
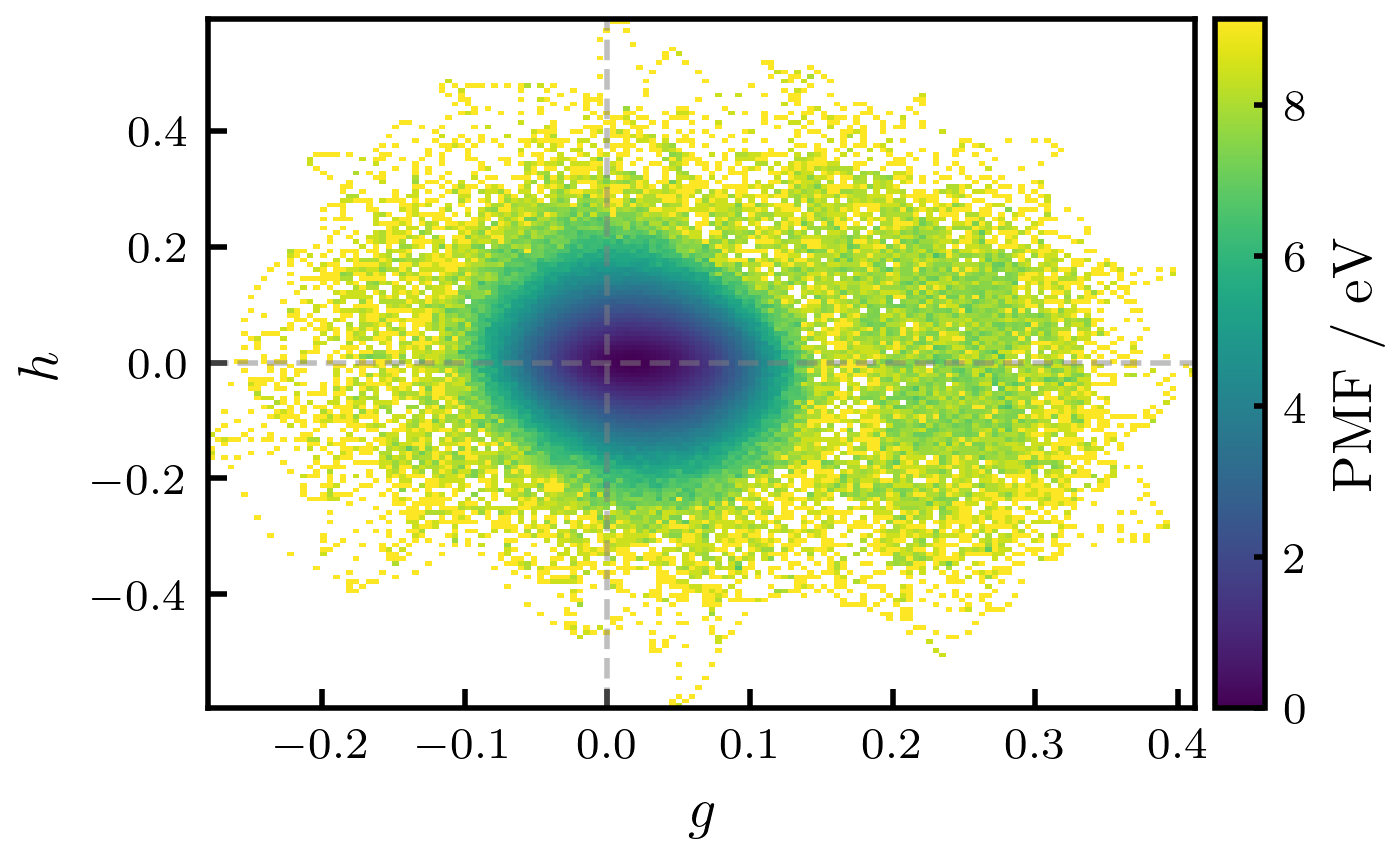
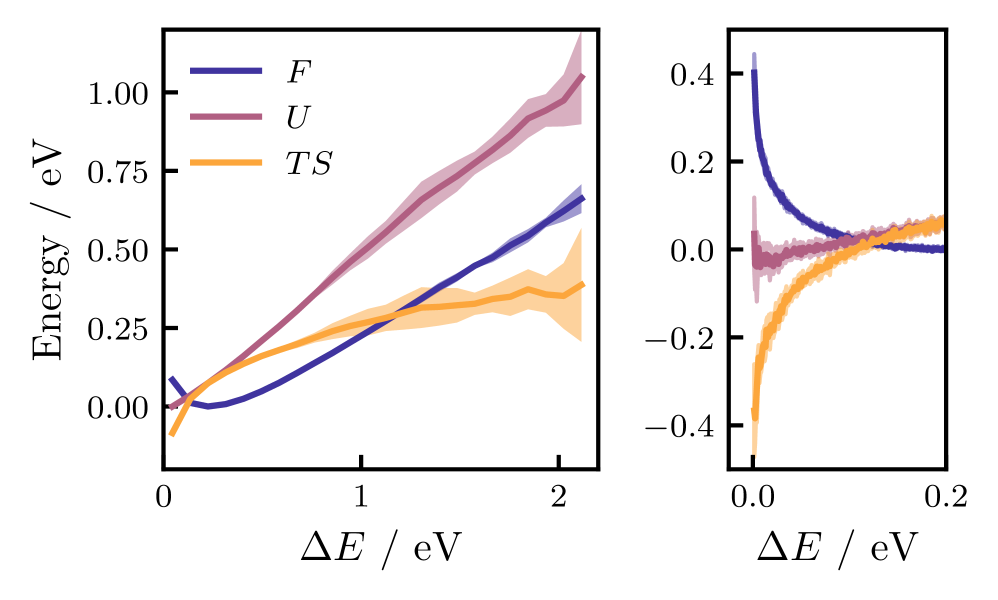
The entropic barrier to accessing the minimum-energy conical intersection in methaniminium cation is a consequence of zero-point vibrational energy and quantum tunneling effects. These effects increase the effective energy required to reach the conical intersection seam, effectively preventing classical trajectories from accessing it. The barrier manifests as a divergence in free energy as the energy gap – a measure of the electronic coupling between states – approaches zero, indicating a statistically improbable transition. This suppression of nonadiabatic transitions occurs because the system must overcome not only the potential energy surface but also the associated vibrational and tunneling contributions to reach the region of minimal energy gap and strong coupling.
Free energy analysis, employing collective variables such as the energy gap between electronic states, is essential for characterizing the entropic barrier influencing nonadiabatic transitions in methaniminium. Calculations demonstrate that classical molecular dynamics simulations fail to access the conical intersection seam due to this barrier; the free energy diverges as the energy gap approaches zero, indicating an infinite barrier height. This divergence arises from the contributions of zero-point vibrational energy and quantum tunneling, effectively precluding statistically significant trajectories from reaching the conical intersection region under classical treatment. The dimensionality of the conical intersection seam is determined to be 3N-8, impacting the complexity of the free energy landscape.
Free energy calculations reveal an infinite potential energy barrier at the conical intersection (CI) seam for methaniminium cation, indicating that classical trajectories are unable to access this region of the potential energy surface. This infinite barrier is not due to a traditional potential energy maximum, but rather arises from the entropic contributions of zero-point vibrational energy and quantum tunneling. Furthermore, the dimensionality of the CI seam itself has been determined to be 3N-8, where N represents the total number of atoms in the molecule; this reduced dimensionality impacts the probability of nonadiabatic transitions and influences the dynamics near the intersection.
Toward Controlling Photochemical Reactions with Precision
Photochemical reactions, fundamental to processes like photosynthesis and vision, often hinge on the precise choreography of molecular motion at conical intersections – points where electronic states meet and nonadiabatic transitions occur. Accurate modeling of these dynamics is therefore not merely a theoretical exercise, but a prerequisite for truly understanding, and ultimately controlling, these reactions. Traditional approaches frequently struggle to capture the full complexity of these events, necessitating advanced computational methods that account for the interplay between electronic and nuclear degrees of freedom. Successfully simulating these dynamics allows researchers to predict reaction pathways, identify key vibrational modes influencing the transition, and explore strategies for manipulating the reaction’s efficiency – potentially leading to innovations in areas like solar energy conversion and the design of novel photoswitches. The ability to navigate these complex potential energy surfaces with precision represents a significant leap towards harnessing the power of light-driven chemistry.
The linear vibronic coupling (LVC) model offers a crucial simplification for understanding how electronic and vibrational states interact at conical intersections – points where potential energy surfaces meet and reactions can occur with remarkable speed. This model posits that the coupling between electronic states is directly proportional to normal vibrational coordinates, effectively treating the interaction as a linear perturbation. V_{el-vib} = \sum_{i} \alpha_{i} Q_{i}, where Q_{i} represents a normal coordinate and \alpha_{i} the coupling coefficient. By focusing on this linear relationship, researchers can analytically and computationally explore the dynamics near conical intersections, predicting how energy flows between electronic and vibrational degrees of freedom and ultimately influencing the outcome of photochemical reactions. The LVC model, while an approximation, provides a powerful framework for interpreting experimental observations and designing strategies for controlling chemical processes with light.
Photochemical reactions, fundamental to processes like photosynthesis and vision, often proceed with low efficiency due to the inherent randomness at conical intersections where electronic states meet. However, recent theoretical work suggests a pathway towards greater control by strategically reshaping the potential energy landscape. This manipulation aims to lower the entropic barrier – the disorder hindering specific reaction pathways – effectively guiding molecules towards desired products. By precisely tailoring the vibrational structure surrounding the conical intersection, researchers envision scenarios where specific vibrational modes are preferentially excited, channeling the reaction along a chosen trajectory and dramatically increasing the yield of target compounds. This approach represents a shift from passively observing photochemical events to actively steering them, potentially revolutionizing fields ranging from materials science to drug discovery.
The study reveals a fundamental limitation in simulating molecular behavior; classical trajectories, despite approaching the conical intersection seam, are effectively barred from reaching it due to an entropic barrier. This echoes Niels Bohr’s assertion, “Every accomplishment comes with a new challenge.” The research demonstrates that even with advanced computational methods, a complete understanding of molecular dynamics requires acknowledging inherent limits-in this case, the inability of classical simulations to fully traverse the potential energy surface. The entropic barrier isn’t merely a technical hurdle, but a reminder that simplification-even in the pursuit of progress-introduces constraints on the completeness of the modeled reality. It is a confirmation that every advancement demands a reassessment of assumptions and limitations.
Beyond the Seam
The demonstration of an entropic barrier preventing traversal to the conical intersection seam reveals a fundamental limitation of applying purely classical intuition to quantum mechanical systems. It is a cautionary reminder that computational efficiency, while valuable, can obscure the very phenomena it seeks to model. The pursuit of faster simulations must not come at the cost of acknowledging the inherent probabilistic nature of molecular dynamics at the quantum level. This is not merely a technical hurdle, but an epistemological one: algorithms, however elegant, are abstractions, and abstractions inevitably lose fidelity.
Future work will undoubtedly focus on refining mixed quantum-classical approaches to circumvent this barrier. However, the persistent challenge lies in defining ‘classical’ in a regime where quantum effects are demonstrably crucial. Simply increasing computational power to sample more trajectories will not resolve the issue; it will only reveal the barrier’s persistence with greater statistical certainty. A deeper investigation into the nature of the degeneracy itself, and how it manifests in the potential energy surface, is required.
Ultimately, the field must confront the possibility that certain quantum phenomena are fundamentally inaccessible to classical simulation, regardless of algorithmic sophistication. Technology without care for people is techno-centrism; similarly, simulation without awareness of its limitations is merely an exercise in self-deception. Ensuring fairness-in this case, accurate representation of physical reality-is part of the engineering discipline.
Original article: https://arxiv.org/pdf/2602.02115.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Gold Rate Forecast
- Looks Like SEGA Is Reheating PS5, PS4 Fan Favourite Sonic Frontiers in Definitive Edition
- Dune 3 Gets the Huge Update Fans Have Been Waiting For
- Pluribus Star Rhea Seehorn Weighs In On That First Kiss
- Kelly Osbourne Slams “Disgusting” Comments on Her Appearance
- Tomodachi Life: Living the Dream ‘Welcome Version’ demo now available
- Action Comics #1096 is Fun Jumping-On Point for Superman Fans (Review)
- Arknights: Endfield – Everything You Need to Know Before You Jump In
- Antiferromagnetic Oscillators: Unlocking Stable Spin Dynamics
- 10 Steamiest Erotic Thriller Movies of the 21st Century
2026-02-03 17:10