Author: Denis Avetisyan
A new study reveals how the quantum behavior of protons in hydrogen-bonded materials subtly alters the way they absorb and emit light.
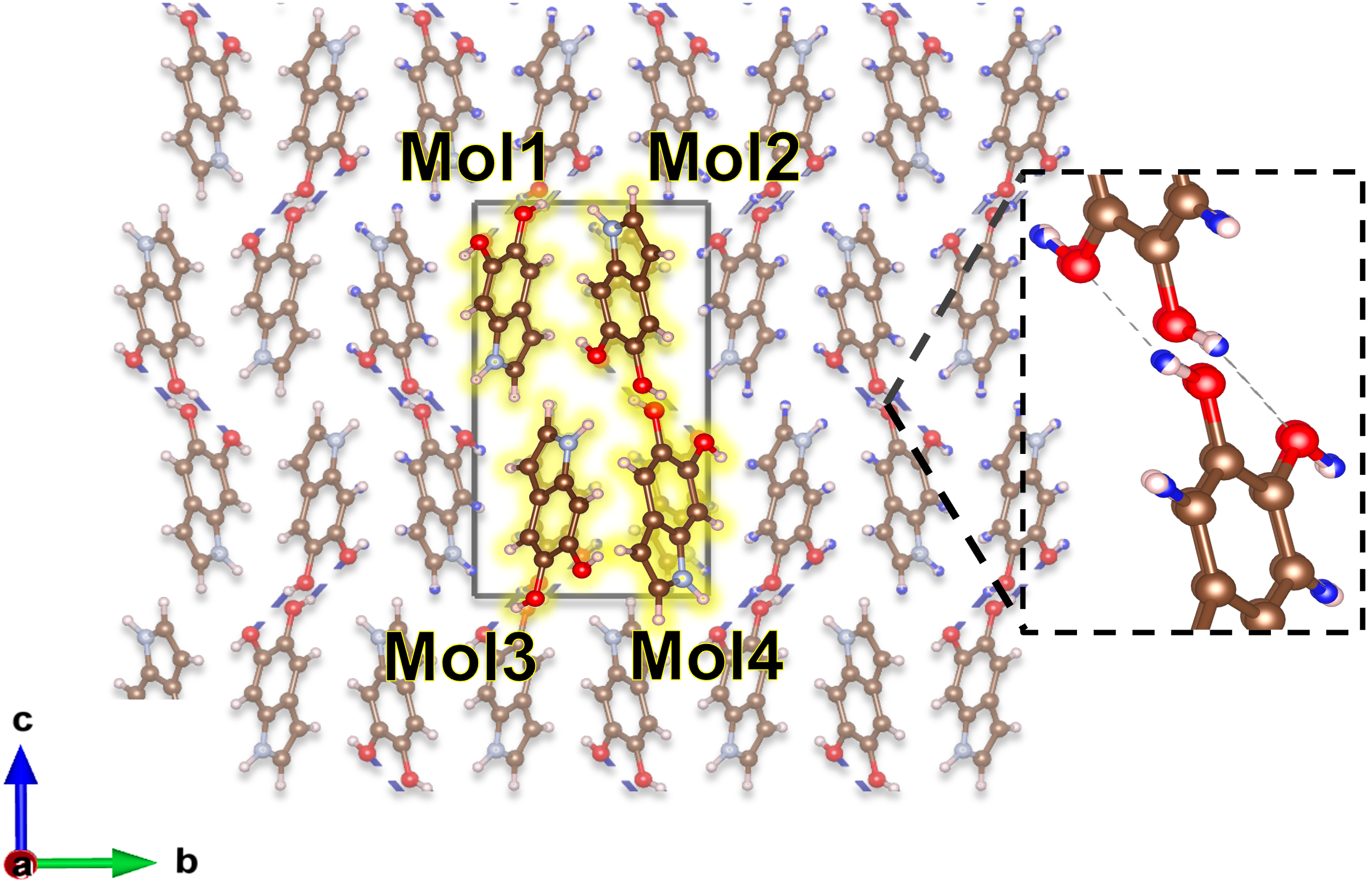
First-principles calculations combining NEO-DFT, G0W0, and the Bethe-Salpeter equation demonstrate the impact of nuclear quantum effects on exciton properties in hydrogen-bonded organic solids.
Despite the known importance of hydrogen bonding in organic materials, the influence of nuclear quantum effects on their excited-state properties remains largely unexplored. This study, ‘Proton Quantum Effects on Electronic Excitation in Hydrogen-bonded Organic Solid: A First-Principles Green’s Function Theory Study’, employs a first-principles Green’s function approach-combining the nuclear-electronic orbital method with the G_0W_0 approximation and Bethe-Salpeter equation-to reveal subtle but significant modifications to exciton behavior in the prototypical hydrogen-bonded solid eumelanin. We demonstrate that proton quantization induces molecular-level anisotropy in excitons and affects their spatial distribution. How might these quantum effects be harnessed to design organic materials with tailored optoelectronic properties?
Eumelanin: A Pigment’s Promise, and a Cautionary Tale
Eumelanin, the pigment responsible for a wide range of colors in skin, hair, and eyes, offers a compelling model for investigating exciton delocalization – the movement of energy within a material. Its structure isn’t simply a rigid framework; instead, it’s characterized by an extensive and intricate network of hydrogen bonds. These bonds, constantly vibrating and shifting, create a dynamic landscape that profoundly influences how excitons – quasi-particles representing excited states of electrons – propagate through the pigment. The complex interplay between the pigment’s molecular arrangement and these hydrogen bonds allows for remarkably efficient energy transfer, potentially protecting against damaging ultraviolet radiation and contributing to the pigment’s broad absorption spectrum. This unique structural feature makes eumelanin a valuable system for understanding fundamental principles of light-matter interaction and exploring applications in bio-inspired materials science.
The optical characteristics of eumelanin, responsible for a wide range of colors in nature, are intimately linked to the precise arrangement of its hydrogen bonds. This network dictates how excitons – energy packets involved in light absorption – move through the pigment’s structure, influencing everything from its brown and black hues to its ability to absorb ultraviolet radiation. Because eumelanin’s hydrogen bonds aren’t static but dynamically fluctuate, even subtle changes in their configuration can dramatically alter the way the pigment interacts with light. Consequently, a detailed understanding of these bonds is not merely academic; it represents a pathway toward controlling eumelanin’s optical properties for potential applications in biomimicry, advanced materials, and even novel photoprotective technologies.
Classical simulations of eumelanin’s hydrogen-bonded network, while providing valuable structural insights, often fall short when predicting its nuanced optical behavior. This limitation arises because these methods treat atoms as fixed points, neglecting the inherent quantum mechanical nature of nuclei – specifically, phenomena like zero-point energy and tunneling. These nuclear quantum effects significantly influence the vibrational modes within the hydrogen bonds, altering the pathways for exciton delocalization and, consequently, the pigment’s absorption and emission properties. Capturing these effects requires computationally demanding approaches, such as path integral molecular dynamics, which account for the probabilistic nature of nuclear motion, offering a more accurate representation of the complex interplay between structure and quantum phenomena within eumelanin.
Excited States: The Devil is in the Details (and the Many-Body Interactions)
The Bethe-Salpeter Equation (BSE) is a many-body perturbation theory approach used to calculate the excitation energies and optical properties of materials. Its accuracy, however, is heavily dependent on the quality of the input quasi-particle energies. These energies are typically obtained from the G_0W_0 approximation, a self-energy correction to the Kohn-Sham eigenvalues of Density Functional Theory (DFT). The G_0W_0 approximation addresses the shortcomings of standard DFT in describing quasi-particle energies by incorporating aspects of many-body interactions, specifically the screened Coulomb interaction W. Errors in the quasi-particle energies, stemming from approximations within the G_0W_0 calculation or limitations of the underlying DFT functional, directly propagate into the BSE results, affecting the predicted excitation energies and oscillator strengths.
Accurate modeling of excited states in systems containing light nuclei, such as those prevalent in the biological pigment eumelanin, requires the inclusion of nuclear quantum effects. Standard electronic structure methods based on the Born-Oppenheimer approximation assume fixed nuclei, which becomes insufficient for light nuclei due to their larger zero-point energy and increased quantum fluctuations. These fluctuations significantly impact the potential energy surface experienced by electrons and, consequently, the calculated excitation energies. Ignoring these effects can lead to substantial errors in predicting optical properties and reactivity. Approaches like Multicomponent Density Functional Theory (MDFT) and the Nuclear-Electronic Orbital (NEO) method address this limitation by explicitly incorporating the quantization of selected nuclear degrees of freedom, thereby providing a more realistic description of the system’s electronic structure and dynamics.
Multicomponent Density Functional Theory (MDFT) addresses the limitations of standard Density Functional Theory (DFT) by explicitly incorporating the quantum mechanical motion of select nuclei, typically light atoms such as hydrogen. Traditional DFT treats nuclei as classical point charges, neglecting zero-point energy and tunneling effects which become significant in systems containing these light nuclei. MDFT achieves this by expanding the wavefunction to include both electronic and nuclear coordinates, effectively quantizing the motion of the chosen nuclei. The Nuclear-Electronic Orbital (NEO) method is a practical implementation of MDFT, employing a basis set expansion suitable for treating both electronic and nuclear degrees of freedom and enabling the calculation of potential energy surfaces that accurately reflect nuclear quantum effects. This approach is crucial for accurately modeling excited states in systems like eumelanin where the motion of protons significantly impacts optical properties.
From Diffraction to Detail: Validating the Model Against Reality
X-ray diffraction was employed to elucidate the three-dimensional arrangement of eumelanin, specifically revealing a helical hydrogen-bonded packing motif. This structural determination is foundational to the computational modeling process, as it provides the precise atomic coordinates and intermolecular interactions necessary for accurate calculations. The observed helical structure influences the electronic properties of the material, dictating the pathways for electron excitation and ultimately impacting its optical characteristics. The diffraction data established the initial geometric parameters used in subsequent quantum mechanical calculations, ensuring that the model reflects the experimentally observed molecular arrangement of eumelanin.
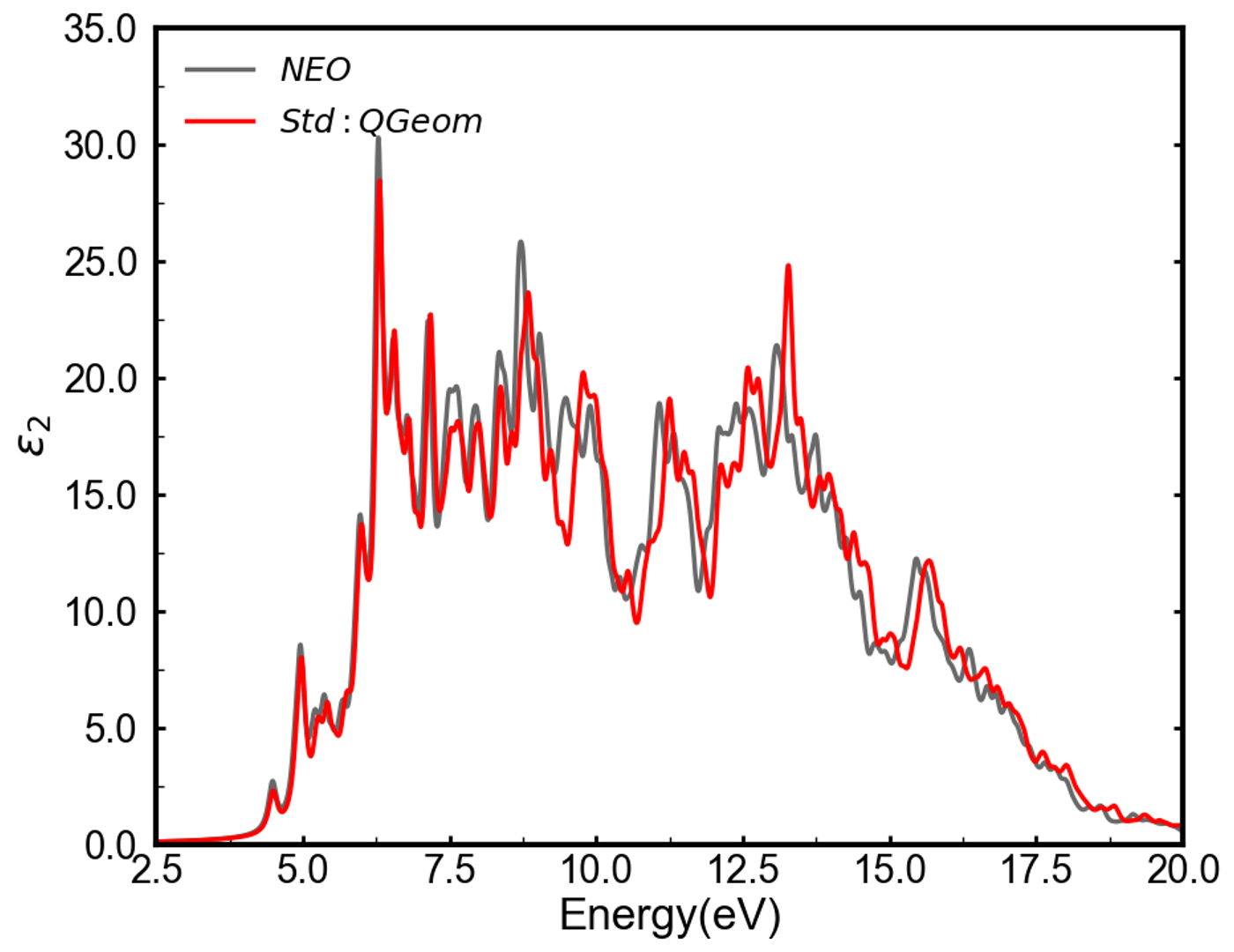
The optical absorption spectrum of eumelanin is accurately modeled through a computational approach combining X-ray diffraction-derived structural information with the NEO-MDFT (New Extended Operator – Many-body Density Functional Theory) methodology. This calculation utilizes a Tier 2 numeric atomic orbital basis set, which provides a balance between computational cost and accuracy in describing the electronic structure. The Tier 2 basis set facilitates the efficient calculation of electronic transitions responsible for the material’s optical properties, enabling a detailed analysis of the absorption spectrum and correlation with experimental data. The NEO-MDFT approach accounts for many-body interactions, crucial for accurately representing the excited state behavior of eumelanin and its characteristic absorption features.
Analysis of the dielectric function and density of states, facilitated by Mulliken population analysis, provides data regarding exciton delocalization within the eumelanin structure and its influence on optical response. Computational results indicate that the inclusion of proton quantum effects leads to a reduction in the quasi-particle energy gap of approximately 0.05 eV. Furthermore, these calculations demonstrate a decrease in exciton binding energy, changing from 1.46 \text{ eV} to 1.41 \text{ eV} when proton quantum effects are considered. These values provide quantitative insight into the material’s electronic structure and its impact on light absorption characteristics.
Beyond Eumelanin: Implications for Materials Design, and a Word of Caution
The design of materials with specific optical characteristics relies heavily on accurately representing the behavior of atomic nuclei, a feat achieved through methods like Nuclear Equation-of-Motion Density Functional Theory (NEO-MDFT). Traditional material modeling often treats atoms as static points, neglecting the inherent quantum nature of nuclei – their motion and associated energy levels. However, these nuclear quantum effects significantly influence how light interacts with a material, affecting its absorption, emission, and overall color. NEO-MDFT addresses this limitation by explicitly incorporating these quantum behaviors into calculations, allowing researchers to predict and tailor optical properties with unprecedented precision. This is particularly crucial for complex materials where hydrogen bonding and proton transfer play a vital role in determining the material’s response to light, ultimately enabling the creation of materials designed for specific applications in areas like solar energy, photonics, and advanced coatings.
Eumelanin, the pigment responsible for dark coloration in skin, hair, and feathers, exhibits remarkably efficient light-harvesting capabilities due to the extensive delocalization of excitons – bound electron-hole pairs that transport energy. Researchers are now leveraging a detailed understanding of this delocalization to inspire the design of novel biomimetic materials. By replicating the structural features that facilitate exciton movement in eumelanin, scientists aim to create synthetic systems capable of capturing and transferring light energy with unprecedented efficiency. These bio-inspired materials hold promise for applications ranging from advanced solar cells and light-sensitive sensors to improved photoprotection and even enhanced optical computing, effectively mimicking nature’s elegant solution to energy transfer challenges.
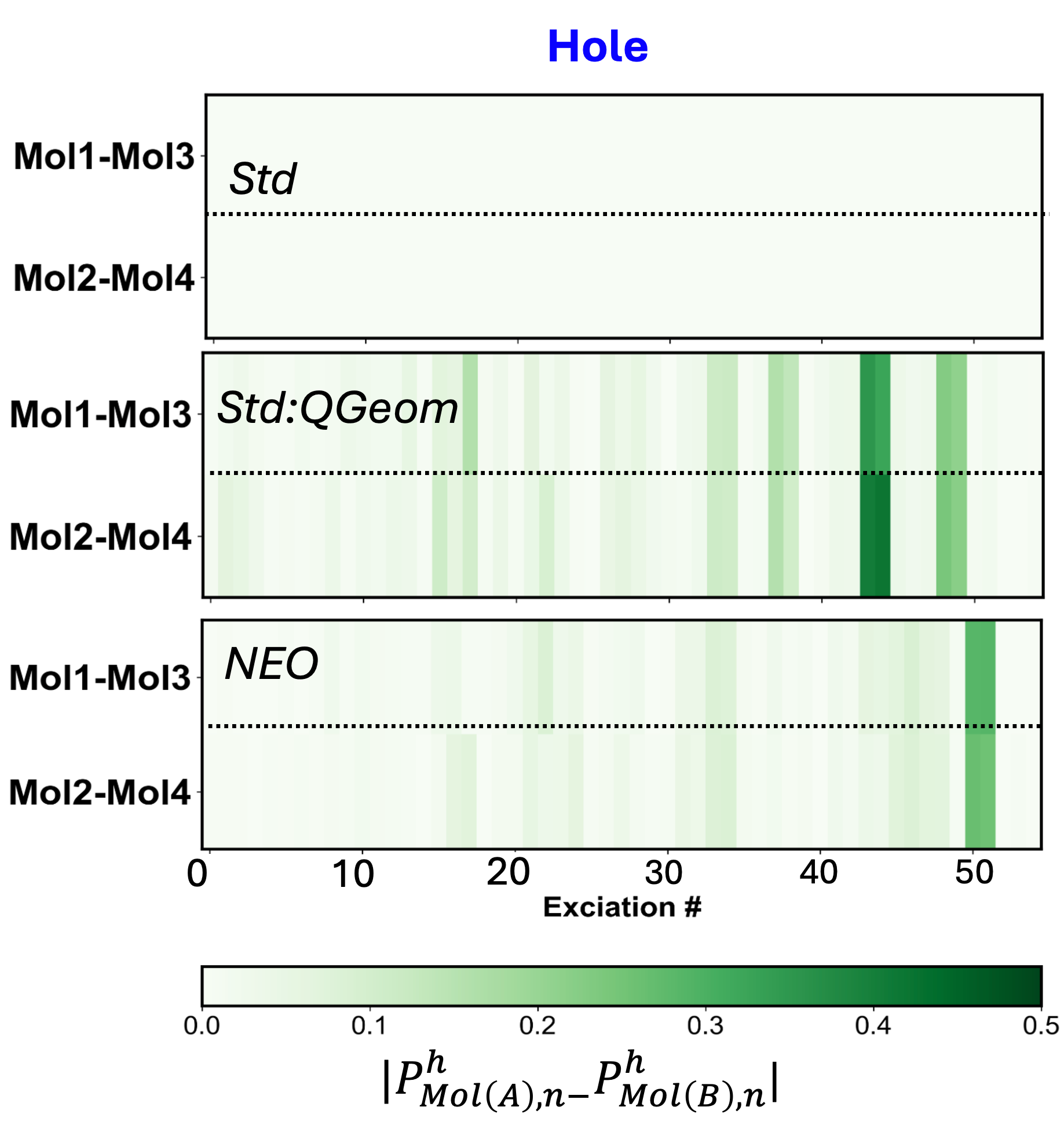
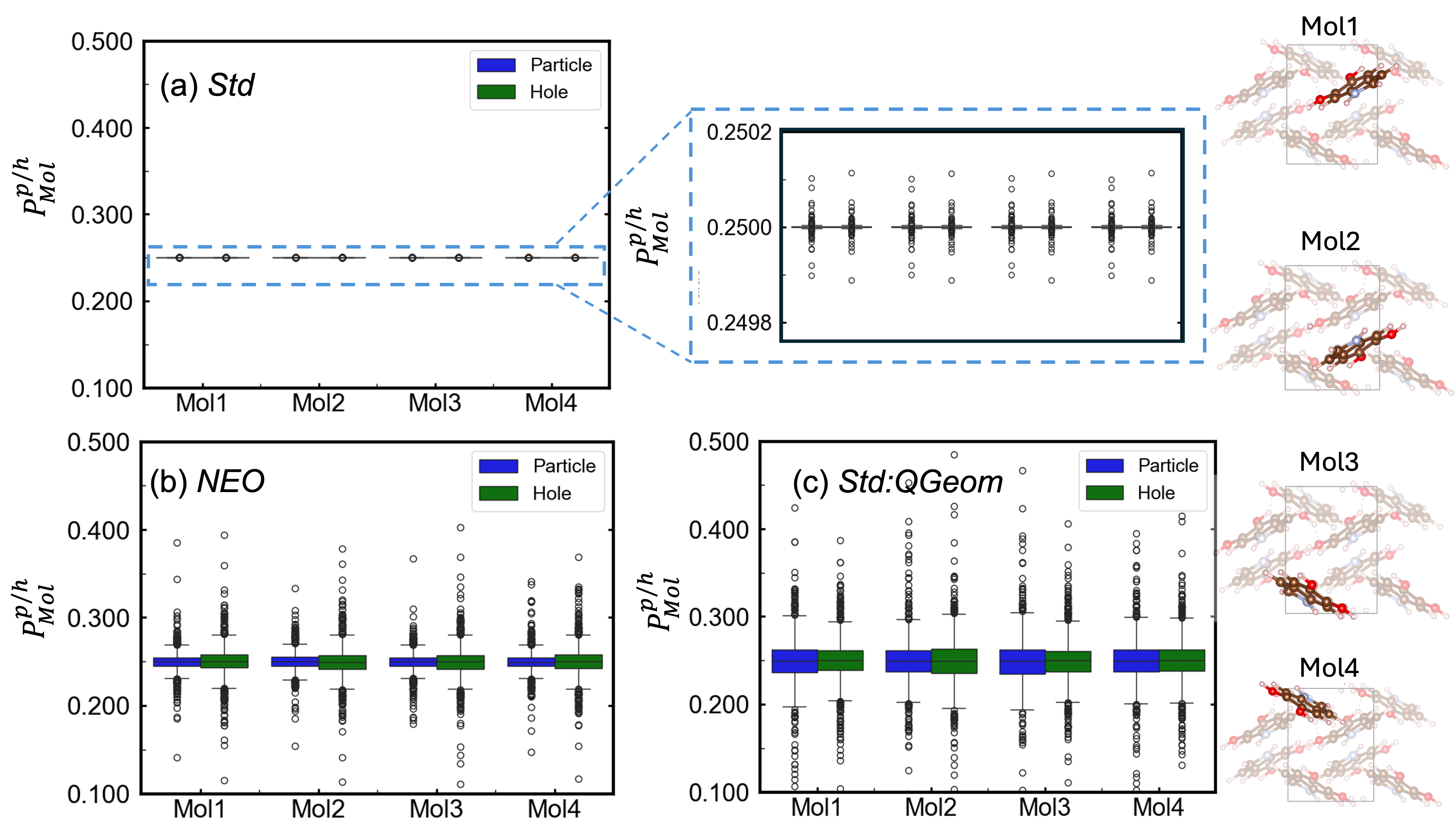
The convergence of advanced theoretical modeling and computational methods establishes a robust platform for dissecting the intricate connections between structure and properties within a diverse array of hydrogen-bonded systems. Investigations utilizing this framework revealed that accounting for proton quantization-a key nuclear quantum effect-substantially alters exciton behavior. Specifically, the standard deviation of Mulliken exciton populations increased by two orders of magnitude upon inclusion of proton quantization, signifying a markedly enhanced exciton anisotropy. This finding suggests that the directional preference of excited states-and thus, optical properties-is significantly more sensitive to quantum mechanical treatment of protons than previously understood, opening avenues for the rational design of materials with tailored light absorption and emission characteristics.
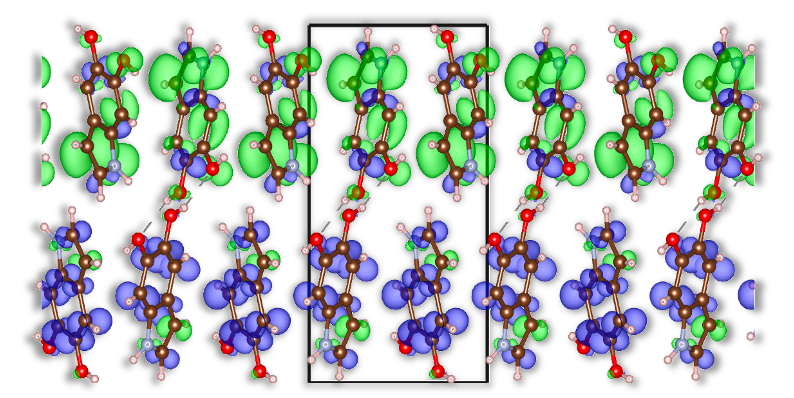
The pursuit of increasingly accurate models, as demonstrated by this study’s layered approach-NEO-DFT, G0W0, and the Bethe-Salpeter equation-feels less like scientific progress and more like exquisitely detailed documentation of inevitable decay. It’s a beautiful edifice built on assumptions that production will, predictably, stress-test into obsolescence. As Aristotle observed, “The ultimate value of life depends upon awareness and the power of contemplation rather than upon mere survival.” This research diligently maps the exciton distribution and anisotropy, but the very act of refinement introduces further abstraction, a complexity that will, in time, become the new technical debt. The more precisely one models reality, the more brittle that model becomes.
So, What Breaks Next?
This exercise in meticulously accounting for proton wiggles – admirable, truly – inevitably bumps up against the limitations of even the most sophisticated theoretical scaffolding. The Bethe-Salpeter equation, while elegant, still relies on approximations. One can refine the functional, tweak the basis sets, and chase ever-smaller numerical noise, but the underlying issue remains: real-world hydrogen bonds aren’t pristine, isolated entities. Defects, impurities, and thermal fluctuations will conspire to dismantle any perfectly predicted exciton distribution. If a system crashes consistently, at least it’s predictable.
The current approach, while illuminating the influence of nuclear quantum effects, doesn’t yet offer a practical pathway to control them. The holy grail-designing organic solids with tailored excitonic properties-remains distant. Future efforts will likely focus on bridging the gap between these first-principles calculations and experimental observables, perhaps through machine learning models trained on increasingly complex datasets. Though, let’s be honest, that’s just trading one set of approximations for another.
It’s a beautiful calculation, certainly. But one suspects that, a decade hence, this NEO-DFT/G0W0/BSE workflow will be viewed as a quaint precursor to whatever the next ‘cloud-native’ quantum chemistry package offers. After all, it’s not about understanding the physics; it’s about generating data for the next grant proposal. And, ultimately, it’s not code that endures-it’s the notes left for digital archaeologists.
Original article: https://arxiv.org/pdf/2602.07791.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Gold Rate Forecast
- Dune 3 Gets the Huge Update Fans Have Been Waiting For
- 22 actors who were almost James Bond – and why they missed out on playing 007
- Hazbin Hotel Secretly Suggests Vox Helped Create One of the Most Infamous Cults in History
- Every Creepy Clown in American Horror Story Ranked
- Jason Statham’s Hit Creature Feature Is Heading to Streaming for Free
- As Dougal and friends turn 60, Radio Times explores the magic behind The Magic Roundabout
- Kingdom Come: Deliverance 2 – Legacy of the Forge DLC Review – Cozy Crafting
- Jack Osbourne Shares Heartbreaking Tribute to Late Dad Ozzy Osbourne
- Everything We Know About Gen V Season 3 (& Why It’ll Be a Very Different Show)
2026-02-11 03:35